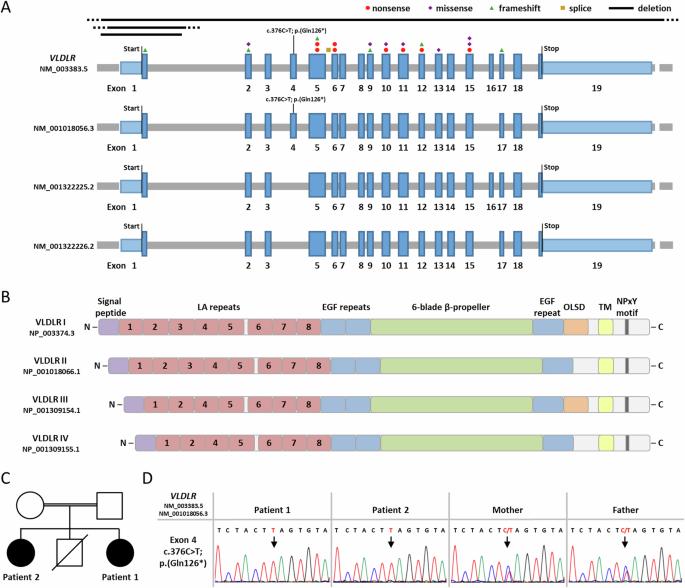
A homozygous nonsense variant in the alternatively spliced VLDLR exon 4 causes a neurodevelopmental disorder without features of VLDLR cerebellar hypoplasia
Tess Holling, Ibrahim M. Abdelrazek, Ghada M. Elhady, Marwa Abd Elmaksoud, Seung Woo Ryu, Ebtesam Abdalla, Kerstin Kutsche
{"title":"A homozygous nonsense variant in the alternatively spliced VLDLR exon 4 causes a neurodevelopmental disorder without features of VLDLR cerebellar hypoplasia","authors":"Tess Holling, Ibrahim M. Abdelrazek, Ghada M. Elhady, Marwa Abd Elmaksoud, Seung Woo Ryu, Ebtesam Abdalla, Kerstin Kutsche","doi":"10.1038/s10038-024-01279-w","DOIUrl":null,"url":null,"abstract":"VLDLR cerebellar hypoplasia is characterized by intellectual disability, non-progressive cerebellar ataxia, and seizures. The characteristic MRI findings include hypoplasia of the inferior portion of the cerebellar vermis and hemispheres, simplified cortical gyration, and a small brain stem. Biallelic VLDLR pathogenic variants cause loss-of-function of the encoded very low-density lipoprotein receptor. VLDLR exons 4 and 16 are alternatively spliced, resulting in the expression of four transcript variants, including two exon 4-lacking mRNAs expressed in the human brain. Previously reported VLDLR pathogenic variants affect all four transcript variants. Here we report on two sisters with facial dysmorphism, microcephaly, intellectual disability, and normal brain imaging. Exome sequencing in one patient identified the homozygous VLDLR nonsense variant c.376C>T; p.(Gln126*) in exon 4; her similarly affected sister also carried the homozygous variant and parents were heterozygous carriers. VLDLR transcript analysis identified mRNAs with and without exon 4 in patient fibroblasts, while exon 4-containing VLDLR mRNAs were predominantly detected in control fibroblasts. We found significantly reduced VLDLR mRNA levels in patient compared to control cells, likely caused by nonsense-mediated mRNA decay of exon 4-containing VLDLR transcripts. Expression of neuronal VLDLR isoforms produced from exon 4-lacking transcripts may have protected both patients from developing the cerebellar hypoplasia phenotype.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 12","pages":"623-628"},"PeriodicalIF":2.5000,"publicationDate":"2024-07-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s10038-024-01279-w.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01279-w","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
VLDLR cerebellar hypoplasia is characterized by intellectual disability, non-progressive cerebellar ataxia, and seizures. The characteristic MRI findings include hypoplasia of the inferior portion of the cerebellar vermis and hemispheres, simplified cortical gyration, and a small brain stem. Biallelic VLDLR pathogenic variants cause loss-of-function of the encoded very low-density lipoprotein receptor. VLDLR exons 4 and 16 are alternatively spliced, resulting in the expression of four transcript variants, including two exon 4-lacking mRNAs expressed in the human brain. Previously reported VLDLR pathogenic variants affect all four transcript variants. Here we report on two sisters with facial dysmorphism, microcephaly, intellectual disability, and normal brain imaging. Exome sequencing in one patient identified the homozygous VLDLR nonsense variant c.376C>T; p.(Gln126*) in exon 4; her similarly affected sister also carried the homozygous variant and parents were heterozygous carriers. VLDLR transcript analysis identified mRNAs with and without exon 4 in patient fibroblasts, while exon 4-containing VLDLR mRNAs were predominantly detected in control fibroblasts. We found significantly reduced VLDLR mRNA levels in patient compared to control cells, likely caused by nonsense-mediated mRNA decay of exon 4-containing VLDLR transcripts. Expression of neuronal VLDLR isoforms produced from exon 4-lacking transcripts may have protected both patients from developing the cerebellar hypoplasia phenotype.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
