{"title":"Antifibrotics and mortality in idiopathic pulmonary fibrosis: external validity and avoidance of immortal time bias.","authors":"Hironao Hozumi, Koichi Miyashita, Eiji Nakatani, Yusuke Inoue, Hideki Yasui, Yuzo Suzuki, Masato Karayama, Kazuki Furuhashi, Noriyuki Enomoto, Tomoyuki Fujisawa, Naoki Inui, Takafumi Suda","doi":"10.1186/s12931-024-02922-y","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objective: </strong>Pooled analyses of previous randomized controlled trials reported that antifibrotics improved survival in patients with idiopathic pulmonary fibrosis (IPF), but the results were only based on short-term outcome data from selected patients who met strict criteria. Observational studies/meta-analyses also suggested that antifibrotics improve survival, but these studies failed to control for immortal time bias that considerably exaggerates drug effects. Therefore, whether antifibrotics truly improve long-term survival in patients with IPF in the real world remains undetermined and requires external validity.</p><p><strong>Methods: </strong>We used data from the Japanese National Claims Database to estimate the intention-to-treat effect of antifibrotics on mortality. To address immortal time bias, we employed models treating antifibrotic initiation as a time-dependent covariate and target trial emulation (TTE), both incorporating new-user designs for antifibrotics and treating lung transplantation as a competing event.</p><p><strong>Results: </strong>Of 30,154 patients with IPF, 14,525 received antifibrotics. Multivariate Fine-Gray models with antifibrotic initiation as a time-dependent covariate revealed that compared with no treatment, nintedanib (adjusted hazard ratio [aHR], 0.85; 95% confidence interval [CI], 0.81-0.89) and pirfenidone (aHR, 0.89; 95% CI, 0.86-0.93) were associated with reduced mortality. The TTE model also replicated the associations of nintedanib (aHR, 0.69; 95% CI, 0.65-0.74) and pirfenidone (aHR, 0.81; 95% CI, 0.78-0.85) with reduced mortality. Subgroup analyses confirmed this association regardless of age, sex, and comorbidities, excluding certain subpopulations.</p><p><strong>Conclusions: </strong>The results of this large-scale real-world analysis support the generalizability of the association between antifibrotics and improved survival in various IPF populations.</p>","PeriodicalId":49131,"journal":{"name":"Respiratory Research","volume":"25 1","pages":"293"},"PeriodicalIF":5.8000,"publicationDate":"2024-07-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11293013/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Respiratory Research","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12931-024-02922-y","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
Abstract
Background and objective: Pooled analyses of previous randomized controlled trials reported that antifibrotics improved survival in patients with idiopathic pulmonary fibrosis (IPF), but the results were only based on short-term outcome data from selected patients who met strict criteria. Observational studies/meta-analyses also suggested that antifibrotics improve survival, but these studies failed to control for immortal time bias that considerably exaggerates drug effects. Therefore, whether antifibrotics truly improve long-term survival in patients with IPF in the real world remains undetermined and requires external validity.
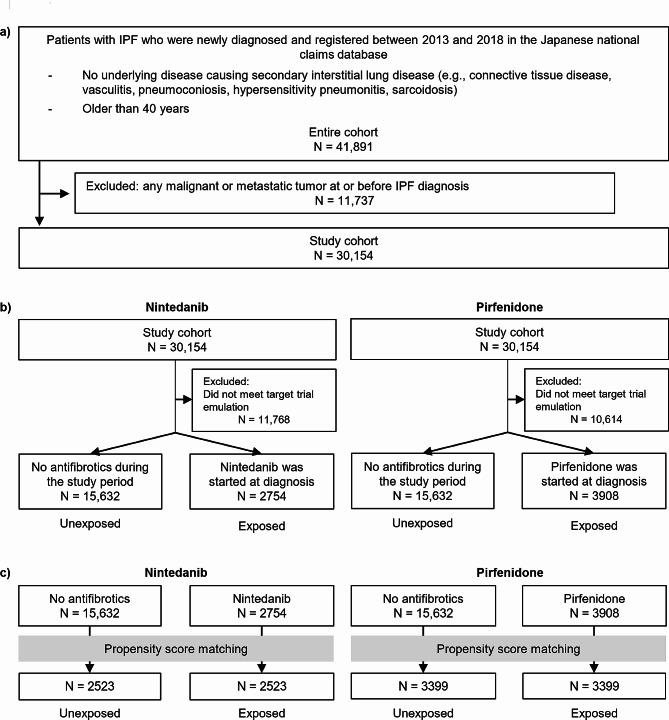
Methods: We used data from the Japanese National Claims Database to estimate the intention-to-treat effect of antifibrotics on mortality. To address immortal time bias, we employed models treating antifibrotic initiation as a time-dependent covariate and target trial emulation (TTE), both incorporating new-user designs for antifibrotics and treating lung transplantation as a competing event.
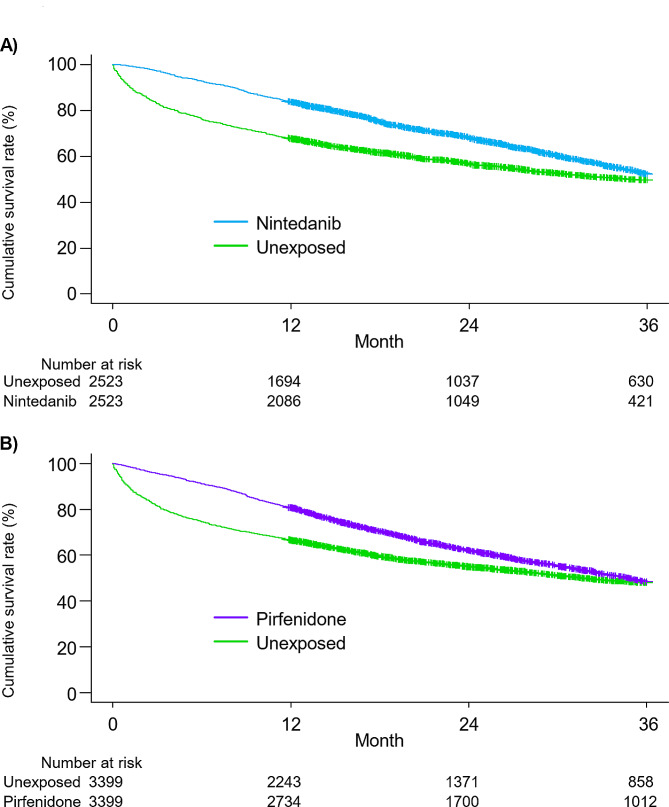
Results: Of 30,154 patients with IPF, 14,525 received antifibrotics. Multivariate Fine-Gray models with antifibrotic initiation as a time-dependent covariate revealed that compared with no treatment, nintedanib (adjusted hazard ratio [aHR], 0.85; 95% confidence interval [CI], 0.81-0.89) and pirfenidone (aHR, 0.89; 95% CI, 0.86-0.93) were associated with reduced mortality. The TTE model also replicated the associations of nintedanib (aHR, 0.69; 95% CI, 0.65-0.74) and pirfenidone (aHR, 0.81; 95% CI, 0.78-0.85) with reduced mortality. Subgroup analyses confirmed this association regardless of age, sex, and comorbidities, excluding certain subpopulations.
Conclusions: The results of this large-scale real-world analysis support the generalizability of the association between antifibrotics and improved survival in various IPF populations.
期刊介绍:
Respiratory Research publishes high-quality clinical and basic research, review and commentary articles on all aspects of respiratory medicine and related diseases.
As the leading fully open access journal in the field, Respiratory Research provides an essential resource for pulmonologists, allergists, immunologists and other physicians, researchers, healthcare workers and medical students with worldwide dissemination of articles resulting in high visibility and generating international discussion.
Topics of specific interest include asthma, chronic obstructive pulmonary disease, cystic fibrosis, genetics, infectious diseases, interstitial lung diseases, lung development, lung tumors, occupational and environmental factors, pulmonary circulation, pulmonary pharmacology and therapeutics, respiratory immunology, respiratory physiology, and sleep-related respiratory problems.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
