{"title":"SMC3 contributes to heart development by regulating super-enhancer associated genes","authors":"Bowen Zhang, Yongchang Zhu, Zhen Zhang, Feizhen Wu, Xiaojing Ma, Wei Sheng, Ranran Dai, Zhenglong Guo, Weili Yan, Lili Hao, Guoying Huang, Duan Ma, Bingtao Hao, Jing Ma","doi":"10.1038/s12276-024-01293-0","DOIUrl":null,"url":null,"abstract":"Abnormal cardiac development has been observed in individuals with Cornelia de Lange syndrome (CdLS) due to mutations in genes encoding members of the cohesin complex. However, the precise role of cohesin in heart development remains elusive. In this study, we aimed to elucidate the indispensable role of SMC3, a component of the cohesin complex, in cardiac development and its underlying mechanism. Our investigation revealed that CdLS patients with SMC3 mutations have high rates of congenital heart disease (CHD). We utilized heart-specific Smc3-knockout (SMC3-cKO) mice, which exhibit varying degrees of outflow tract (OFT) abnormalities, to further explore this relationship. Additionally, we identified 16 rare SMC3 variants with potential pathogenicity in individuals with isolated CHD. By employing single-nucleus RNA sequencing and chromosome conformation capture high-throughput genome-wide translocation sequencing, we revealed that Smc3 deletion downregulates the expression of key genes, including Ets2, in OFT cardiac muscle cells by specifically decreasing interactions between super-enhancers (SEs) and promoters. Notably, Ets2-SE-null mice also exhibit delayed OFT development in the heart. Our research revealed a novel role for SMC3 in heart development via the regulation of SE-associated genes, suggesting its potential relevance as a CHD-related gene and providing crucial insights into the molecular basis of cardiac development. Understanding heart development is vital as defects in this process are a major cause of birth abnormalities. This study focuses on a protein, SMC3, and its role in heart development. Experiments were conducted on mice genetically altered to lack SMC3 in heart cells. Researchers found that mice without SMC3 had various heart defects, like those seen in humans with congenital heart disease. They also found mutations in the SMC3 gene in patients with congenital heart disease, suggesting a link between SMC3 and heart development in humans. The findings reveal that SMC3 plays a crucial role in heart development, with its absence leading to significant heart defects in mice. These results suggest a potential genetic cause for some forms of congenital heart disease in humans. This summary was initially drafted using artificial intelligence, then revised and fact-checked by the author. Introduction","PeriodicalId":50466,"journal":{"name":"Experimental and Molecular Medicine","volume":"56 8","pages":"1826-1842"},"PeriodicalIF":12.9000,"publicationDate":"2024-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s12276-024-01293-0.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Experimental and Molecular Medicine","FirstCategoryId":"3","ListUrlMain":"https://www.nature.com/articles/s12276-024-01293-0","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
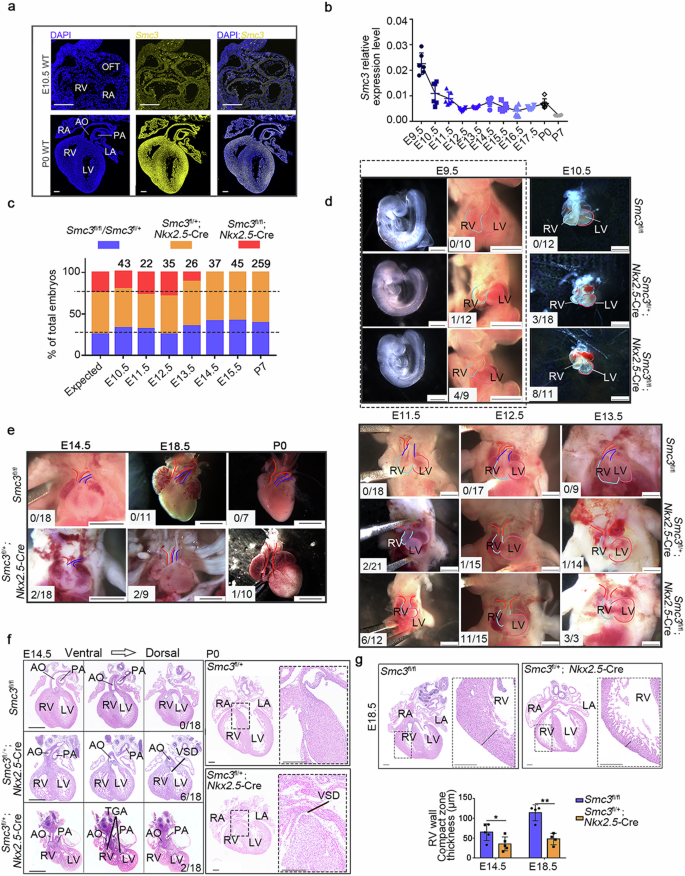
Abnormal cardiac development has been observed in individuals with Cornelia de Lange syndrome (CdLS) due to mutations in genes encoding members of the cohesin complex. However, the precise role of cohesin in heart development remains elusive. In this study, we aimed to elucidate the indispensable role of SMC3, a component of the cohesin complex, in cardiac development and its underlying mechanism. Our investigation revealed that CdLS patients with SMC3 mutations have high rates of congenital heart disease (CHD). We utilized heart-specific Smc3-knockout (SMC3-cKO) mice, which exhibit varying degrees of outflow tract (OFT) abnormalities, to further explore this relationship. Additionally, we identified 16 rare SMC3 variants with potential pathogenicity in individuals with isolated CHD. By employing single-nucleus RNA sequencing and chromosome conformation capture high-throughput genome-wide translocation sequencing, we revealed that Smc3 deletion downregulates the expression of key genes, including Ets2, in OFT cardiac muscle cells by specifically decreasing interactions between super-enhancers (SEs) and promoters. Notably, Ets2-SE-null mice also exhibit delayed OFT development in the heart. Our research revealed a novel role for SMC3 in heart development via the regulation of SE-associated genes, suggesting its potential relevance as a CHD-related gene and providing crucial insights into the molecular basis of cardiac development. Understanding heart development is vital as defects in this process are a major cause of birth abnormalities. This study focuses on a protein, SMC3, and its role in heart development. Experiments were conducted on mice genetically altered to lack SMC3 in heart cells. Researchers found that mice without SMC3 had various heart defects, like those seen in humans with congenital heart disease. They also found mutations in the SMC3 gene in patients with congenital heart disease, suggesting a link between SMC3 and heart development in humans. The findings reveal that SMC3 plays a crucial role in heart development, with its absence leading to significant heart defects in mice. These results suggest a potential genetic cause for some forms of congenital heart disease in humans. This summary was initially drafted using artificial intelligence, then revised and fact-checked by the author. Introduction
期刊介绍:
Experimental & Molecular Medicine (EMM) stands as Korea's pioneering biochemistry journal, established in 1964 and rejuvenated in 1996 as an Open Access, fully peer-reviewed international journal. Dedicated to advancing translational research and showcasing recent breakthroughs in the biomedical realm, EMM invites submissions encompassing genetic, molecular, and cellular studies of human physiology and diseases. Emphasizing the correlation between experimental and translational research and enhanced clinical benefits, the journal actively encourages contributions employing specific molecular tools. Welcoming studies that bridge basic discoveries with clinical relevance, alongside articles demonstrating clear in vivo significance and novelty, Experimental & Molecular Medicine proudly serves as an open-access, online-only repository of cutting-edge medical research.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
