Michele Nottoli, Michael F. Herbst, Aleksandr Mikhalev, Abhinav Jha, Filippo Lipparini, Benjamin Stamm
{"title":"ddX: Polarizable continuum solvation from small molecules to proteins","authors":"Michele Nottoli, Michael F. Herbst, Aleksandr Mikhalev, Abhinav Jha, Filippo Lipparini, Benjamin Stamm","doi":"10.1002/wcms.1726","DOIUrl":null,"url":null,"abstract":"<p>Polarizable continuum solvation models are popular in both, quantum chemistry and in biophysics, though typically with different requirements for the numerical methods. However, the recent trend of multiscale modeling can be expected to blur field-specific differences. In this regard, numerical methods based on domain decomposition (dd) have been demonstrated to be sufficiently flexible to be applied all across these levels of theory while remaining systematically accurate and efficient. In this contribution, we present <span>ddX</span>, an open-source implementation of dd-methods for various solvation models, which features a uniform interface with classical as well as quantum descriptions of the solute, or any hybrid versions thereof. We explain the key concepts of the library design and its application program interface, and demonstrate the use of <span>ddX</span> for integrating into standard chemistry packages. Numerical tests illustrate the performance of <span>ddX</span> and its interfaces.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"14 4","pages":""},"PeriodicalIF":27.0000,"publicationDate":"2024-07-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1726","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1726","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
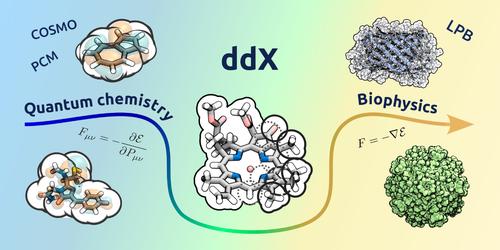
Polarizable continuum solvation models are popular in both, quantum chemistry and in biophysics, though typically with different requirements for the numerical methods. However, the recent trend of multiscale modeling can be expected to blur field-specific differences. In this regard, numerical methods based on domain decomposition (dd) have been demonstrated to be sufficiently flexible to be applied all across these levels of theory while remaining systematically accurate and efficient. In this contribution, we present ddX, an open-source implementation of dd-methods for various solvation models, which features a uniform interface with classical as well as quantum descriptions of the solute, or any hybrid versions thereof. We explain the key concepts of the library design and its application program interface, and demonstrate the use of ddX for integrating into standard chemistry packages. Numerical tests illustrate the performance of ddX and its interfaces.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.






 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
