Andrés Vicente-Acosta , Saúl Herranz-Martín , Maria Ruth Pazos , Jorge Galán-Cruz , Mario Amores , Frida Loria , Javier Díaz-Nido
{"title":"Glial cell activation precedes neurodegeneration in the cerebellar cortex of the YG8–800 murine model of Friedreich ataxia","authors":"Andrés Vicente-Acosta , Saúl Herranz-Martín , Maria Ruth Pazos , Jorge Galán-Cruz , Mario Amores , Frida Loria , Javier Díaz-Nido","doi":"10.1016/j.nbd.2024.106631","DOIUrl":null,"url":null,"abstract":"<div><p>Friedreich ataxia is a hereditary neurodegenerative disorder resulting from reduced levels of the protein frataxin due to an expanded GAA repeat in the <em>FXN</em> gene. This deficiency causes progressive degeneration of specific neuronal populations in the cerebellum and the consequent loss of movement coordination and equilibrium, which are some of the main symptoms observed in affected individuals. Like in other neurodegenerative diseases, previous studies suggest that glial cells could be involved in the neurodegenerative process and disease progression in patients with Friedreich ataxia.</p><p>In this work, we followed and characterized the progression of changes in the cerebellar cortex in the latest version of Friedreich ataxia humanized mouse model, YG8–800 (Fxn<sup>null</sup>:YG8s(GAA)<sub>>800</sub>), which carries a human <em>FXN</em> transgene containing >800 GAA repeats.</p><p>Comparative analyses of behavioral, histopathological, and biochemical parameters were conducted between the control strain Y47R and YG8–800 mice at different time points. Our findings revealed that YG8–800 mice exhibit an ataxic phenotype characterized by poor motor coordination, decreased body weight, cerebellar atrophy, neuronal loss, and changes in synaptic proteins. Additionally, early activation of glial cells, predominantly astrocytes and microglia, was observed preceding neuronal degeneration, as was increased expression of key proinflammatory cytokines and downregulation of neurotrophic factors.</p><p>Together, our results show that the YG8–800 mouse model exhibits a stronger phenotype than previous experimental murine models, reliably recapitulating some of the features observed in humans. Accordingly, this humanized model could represent a valuable tool for studying Friedreich ataxia molecular disease mechanisms and for preclinical evaluation of possible therapies.</p></div>","PeriodicalId":19097,"journal":{"name":"Neurobiology of Disease","volume":"200 ","pages":"Article 106631"},"PeriodicalIF":5.6000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S0969996124002316/pdfft?md5=826328ae0cfb03c076f5f97707d517b9&pid=1-s2.0-S0969996124002316-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurobiology of Disease","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0969996124002316","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/5 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
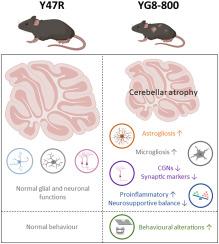
Friedreich ataxia is a hereditary neurodegenerative disorder resulting from reduced levels of the protein frataxin due to an expanded GAA repeat in the FXN gene. This deficiency causes progressive degeneration of specific neuronal populations in the cerebellum and the consequent loss of movement coordination and equilibrium, which are some of the main symptoms observed in affected individuals. Like in other neurodegenerative diseases, previous studies suggest that glial cells could be involved in the neurodegenerative process and disease progression in patients with Friedreich ataxia.
In this work, we followed and characterized the progression of changes in the cerebellar cortex in the latest version of Friedreich ataxia humanized mouse model, YG8–800 (Fxnnull:YG8s(GAA)>800), which carries a human FXN transgene containing >800 GAA repeats.
Comparative analyses of behavioral, histopathological, and biochemical parameters were conducted between the control strain Y47R and YG8–800 mice at different time points. Our findings revealed that YG8–800 mice exhibit an ataxic phenotype characterized by poor motor coordination, decreased body weight, cerebellar atrophy, neuronal loss, and changes in synaptic proteins. Additionally, early activation of glial cells, predominantly astrocytes and microglia, was observed preceding neuronal degeneration, as was increased expression of key proinflammatory cytokines and downregulation of neurotrophic factors.
Together, our results show that the YG8–800 mouse model exhibits a stronger phenotype than previous experimental murine models, reliably recapitulating some of the features observed in humans. Accordingly, this humanized model could represent a valuable tool for studying Friedreich ataxia molecular disease mechanisms and for preclinical evaluation of possible therapies.
期刊介绍:
Neurobiology of Disease is a major international journal at the interface between basic and clinical neuroscience. The journal provides a forum for the publication of top quality research papers on: molecular and cellular definitions of disease mechanisms, the neural systems and underpinning behavioral disorders, the genetics of inherited neurological and psychiatric diseases, nervous system aging, and findings relevant to the development of new therapies.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
