Marcus A Mall, Pierre-Régis Burgel, Carlo Castellani, Jane C Davies, Matthias Salathe, Jennifer L Taylor-Cousar
{"title":"Cystic fibrosis.","authors":"Marcus A Mall, Pierre-Régis Burgel, Carlo Castellani, Jane C Davies, Matthias Salathe, Jennifer L Taylor-Cousar","doi":"10.1038/s41572-024-00538-6","DOIUrl":null,"url":null,"abstract":"<p><p>Cystic fibrosis is a rare genetic disease caused by mutations in CFTR, the gene encoding cystic fibrosis transmembrane conductance regulator (CFTR). The discovery of CFTR in 1989 has enabled the unravelling of disease mechanisms and, more recently, the development of CFTR-directed therapeutics that target the underlying molecular defect. The CFTR protein functions as an ion channel that is crucial for correct ion and fluid transport across epithelial cells lining the airways and other organs. Consequently, CFTR dysfunction causes a complex multi-organ disease but, to date, most of the morbidity and mortality in people with cystic fibrosis is due to muco-obstructive lung disease. Cystic fibrosis care has long been limited to treating symptoms using nutritional support, airway clearance techniques and antibiotics to suppress airway infection. The widespread implementation of newborn screening for cystic fibrosis and the introduction of a highly effective triple combination CFTR modulator therapy that has unprecedented clinical benefits in up to 90% of genetically eligible people with cystic fibrosis has fundamentally changed the therapeutic landscape and improved prognosis. However, people with cystic fibrosis who are not eligible based on their CFTR genotype or who live in countries where they do not have access to this breakthrough therapy remain with a high unmet medical need.</p>","PeriodicalId":18910,"journal":{"name":"Nature Reviews Disease Primers","volume":"10 1","pages":"53"},"PeriodicalIF":76.9000,"publicationDate":"2024-08-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Reviews Disease Primers","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41572-024-00538-6","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
Abstract
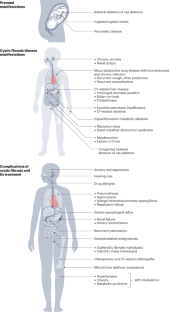
Cystic fibrosis is a rare genetic disease caused by mutations in CFTR, the gene encoding cystic fibrosis transmembrane conductance regulator (CFTR). The discovery of CFTR in 1989 has enabled the unravelling of disease mechanisms and, more recently, the development of CFTR-directed therapeutics that target the underlying molecular defect. The CFTR protein functions as an ion channel that is crucial for correct ion and fluid transport across epithelial cells lining the airways and other organs. Consequently, CFTR dysfunction causes a complex multi-organ disease but, to date, most of the morbidity and mortality in people with cystic fibrosis is due to muco-obstructive lung disease. Cystic fibrosis care has long been limited to treating symptoms using nutritional support, airway clearance techniques and antibiotics to suppress airway infection. The widespread implementation of newborn screening for cystic fibrosis and the introduction of a highly effective triple combination CFTR modulator therapy that has unprecedented clinical benefits in up to 90% of genetically eligible people with cystic fibrosis has fundamentally changed the therapeutic landscape and improved prognosis. However, people with cystic fibrosis who are not eligible based on their CFTR genotype or who live in countries where they do not have access to this breakthrough therapy remain with a high unmet medical need.
期刊介绍:
Nature Reviews Disease Primers, a part of the Nature Reviews journal portfolio, features sections on epidemiology, mechanisms, diagnosis, management, and patient quality of life. The editorial team commissions top researchers — comprising basic scientists and clinical researchers — to write the Primers, which are designed for use by early career researchers, medical students and principal investigators. Each Primer concludes with an Outlook section, highlighting future research directions. Covered medical specialties include Cardiology, Dermatology, Ear, Nose and Throat, Emergency Medicine, Endocrinology, Gastroenterology, Genetic Conditions, Gynaecology and Obstetrics, Hepatology, Haematology, Infectious Diseases, Maxillofacial and Oral Medicine, Nephrology, Neurology, Nutrition, Oncology, Ophthalmology, Orthopaedics, Psychiatry, Respiratory Medicine, Rheumatology, Sleep Medicine, and Urology.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
