Zhili Li, Woojun Kim, Sagar Utturkar, Bingyu Yan, Nadia Atallah Lanman, Bennett D Elzey, Majid Kazemian, Yoon Yeo, Ourania Andrisani
{"title":"DDX5 deficiency drives non-canonical NF-κB activation and NRF2 expression, influencing sorafenib response and hepatocellular carcinoma progression.","authors":"Zhili Li, Woojun Kim, Sagar Utturkar, Bingyu Yan, Nadia Atallah Lanman, Bennett D Elzey, Majid Kazemian, Yoon Yeo, Ourania Andrisani","doi":"10.1038/s41419-024-06977-z","DOIUrl":null,"url":null,"abstract":"<p><p>In advanced hepatocellular carcinoma (HCC), RNA helicase DDX5 regulates the Wnt/β-catenin-ferroptosis axis, influencing the efficacy of the multi-tyrosine kinase inhibitor (mTKI) sorafenib. DDX5 inhibits Wnt/β-catenin signaling, preventing sorafenib-induced ferroptosis escape. Sorafenib/mTKIs reduce DDX5 expression, correlating with poor patient survival post-sorafenib treatment. Notably, DDX5-knockout in HCC cells activates Wnt/β-catenin signaling persistently. Herein, we investigate the mechanistic impact of Wnt/β-catenin activation resulting from DDX5 downregulation in the progression and treatment of HCC. RNAseq analyses identified shared genes repressed by DDX5 and upregulated by sorafenib, including Wnt signaling genes, NF-κB-inducing kinase (NIK) essential for non-canonical NF-κB (p52/RelB) activation, and cytoprotective transcription factor NRF2. We demonstrate, Wnt/β-catenin activation induced NIK transcription, leading to non-canonical NF-κB activation, which subsequently mediated NRF2 transcription. Additionally, DDX5 deficiency extended NRF2 protein half-life by inactivating KEAP1 through p62/SQSTM1 stabilization. In a preclinical HCC mouse model, NRF2 knockdown or DDX5 overexpression restricted tumor growth upon sorafenib treatment, via induction of ferroptosis. Importantly, DDX5-knockout HCC cells exhibited elevated expression of Wnt signaling genes, NIK, p52/RelB, and NRF2-regulated genes, regardless of sorafenib treatment. Transcriptomic analyses of HCCs from TCGA and the Stelic Animal Model (STAM) of non-alcoholic steatohepatitis revealed elevated expression of these interconnected pathways in the context of DDX5 downregulation. In conclusion, DDX5 deficiency triggers Wnt/β-catenin signaling, promoting p52/RelB and NRF2 activation, thereby enabling ferroptosis evasion upon sorafenib treatment. Similarly, independent of sorafenib, DDX5 deficiency in liver tumors enhances activation and gene expression of these interconnected pathways, underscoring the clinical relevance of DDX5 deficiency in HCC progression and therapeutic response.</p>","PeriodicalId":9734,"journal":{"name":"Cell Death & Disease","volume":"15 8","pages":"583"},"PeriodicalIF":9.6000,"publicationDate":"2024-08-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11315975/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell Death & Disease","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1038/s41419-024-06977-z","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
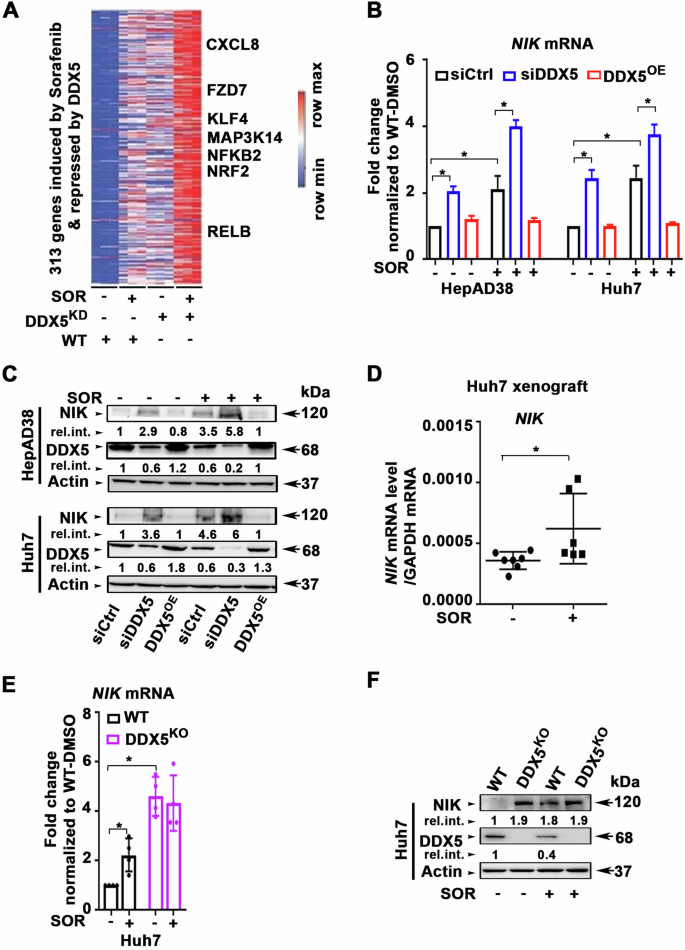
In advanced hepatocellular carcinoma (HCC), RNA helicase DDX5 regulates the Wnt/β-catenin-ferroptosis axis, influencing the efficacy of the multi-tyrosine kinase inhibitor (mTKI) sorafenib. DDX5 inhibits Wnt/β-catenin signaling, preventing sorafenib-induced ferroptosis escape. Sorafenib/mTKIs reduce DDX5 expression, correlating with poor patient survival post-sorafenib treatment. Notably, DDX5-knockout in HCC cells activates Wnt/β-catenin signaling persistently. Herein, we investigate the mechanistic impact of Wnt/β-catenin activation resulting from DDX5 downregulation in the progression and treatment of HCC. RNAseq analyses identified shared genes repressed by DDX5 and upregulated by sorafenib, including Wnt signaling genes, NF-κB-inducing kinase (NIK) essential for non-canonical NF-κB (p52/RelB) activation, and cytoprotective transcription factor NRF2. We demonstrate, Wnt/β-catenin activation induced NIK transcription, leading to non-canonical NF-κB activation, which subsequently mediated NRF2 transcription. Additionally, DDX5 deficiency extended NRF2 protein half-life by inactivating KEAP1 through p62/SQSTM1 stabilization. In a preclinical HCC mouse model, NRF2 knockdown or DDX5 overexpression restricted tumor growth upon sorafenib treatment, via induction of ferroptosis. Importantly, DDX5-knockout HCC cells exhibited elevated expression of Wnt signaling genes, NIK, p52/RelB, and NRF2-regulated genes, regardless of sorafenib treatment. Transcriptomic analyses of HCCs from TCGA and the Stelic Animal Model (STAM) of non-alcoholic steatohepatitis revealed elevated expression of these interconnected pathways in the context of DDX5 downregulation. In conclusion, DDX5 deficiency triggers Wnt/β-catenin signaling, promoting p52/RelB and NRF2 activation, thereby enabling ferroptosis evasion upon sorafenib treatment. Similarly, independent of sorafenib, DDX5 deficiency in liver tumors enhances activation and gene expression of these interconnected pathways, underscoring the clinical relevance of DDX5 deficiency in HCC progression and therapeutic response.
期刊介绍:
Brought to readers by the editorial team of Cell Death & Differentiation, Cell Death & Disease is an online peer-reviewed journal specializing in translational cell death research. It covers a wide range of topics in experimental and internal medicine, including cancer, immunity, neuroscience, and now cancer metabolism.
Cell Death & Disease seeks to encompass the breadth of translational implications of cell death, and topics of particular concentration will include, but are not limited to, the following:
Experimental medicine
Cancer
Immunity
Internal medicine
Neuroscience
Cancer metabolism



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
