{"title":"Exploring unsolved cases of lissencephaly spectrum: integrating exome and genome sequencing for higher diagnostic yield","authors":"Shogo Furukawa, Mitsuhiro Kato, Akihiko Ishiyama, Tomohiro Kumada, Takeshi Yoshida, Eri Takeshita, Pin Fee Chong, Hideo Yamanouchi, Yuko Kotake, Takayoshi Kyoda, Toshihiro Nomura, Yohane Miyata, Mitsuko Nakashima, Hirotomo Saitsu","doi":"10.1038/s10038-024-01283-0","DOIUrl":null,"url":null,"abstract":"Lissencephaly is a rare brain malformation characterized by abnormal neuronal migration during cortical development. In this study, we performed a comprehensive genetic analysis using next-generation sequencing in 12 unsolved Japanese lissencephaly patients, in whom PAFAH1B1, DCX, TUBA1A, and ARX variants were excluded using the Sanger method. Exome sequencing (ES) was conducted on these 12 patients, identifying pathogenic variants in CEP85L, DYNC1H1, LAMC3, and DCX in four patients. Next, we performed genome sequencing (GS) on eight unsolved patients, and structural variants in PAFAH1B1, including an inversion and microdeletions involving several exons, were detected in three patients. Notably, these microdeletions in PAFAH1B1 could not to be detected by copy number variation (CNV) detection tools based on the depth of coverage methods using ES data. The density of repeat sequences, including Alu sequences or segmental duplications, which increase the susceptibility to structural variations, is very high in some lissencephaly spectrum genes (PAFAH1B1, TUBA1A, DYNC1H1). These missing CNVs were due to the limitations of detecting repeat sequences in ES-based CNV detection tools. Our study suggests that a combined approach integrating ES with GS can contribute to a higher diagnostic yield and a better understanding of the genetic landscape of the lissencephaly spectrum.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"69 12","pages":"629-637"},"PeriodicalIF":2.5000,"publicationDate":"2024-08-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01283-0","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
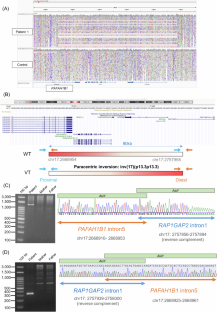
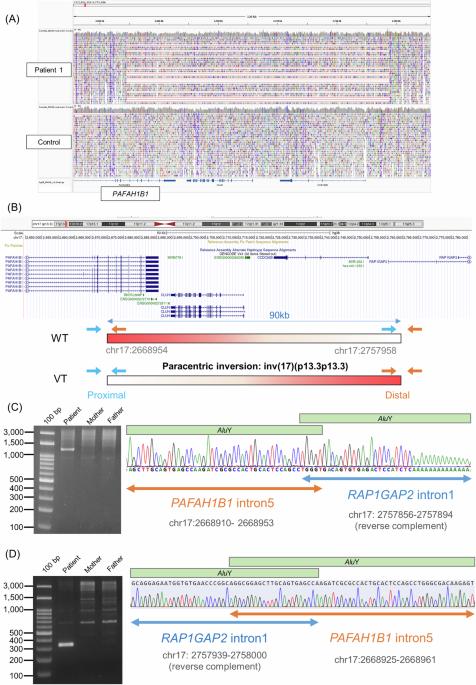
Lissencephaly is a rare brain malformation characterized by abnormal neuronal migration during cortical development. In this study, we performed a comprehensive genetic analysis using next-generation sequencing in 12 unsolved Japanese lissencephaly patients, in whom PAFAH1B1, DCX, TUBA1A, and ARX variants were excluded using the Sanger method. Exome sequencing (ES) was conducted on these 12 patients, identifying pathogenic variants in CEP85L, DYNC1H1, LAMC3, and DCX in four patients. Next, we performed genome sequencing (GS) on eight unsolved patients, and structural variants in PAFAH1B1, including an inversion and microdeletions involving several exons, were detected in three patients. Notably, these microdeletions in PAFAH1B1 could not to be detected by copy number variation (CNV) detection tools based on the depth of coverage methods using ES data. The density of repeat sequences, including Alu sequences or segmental duplications, which increase the susceptibility to structural variations, is very high in some lissencephaly spectrum genes (PAFAH1B1, TUBA1A, DYNC1H1). These missing CNVs were due to the limitations of detecting repeat sequences in ES-based CNV detection tools. Our study suggests that a combined approach integrating ES with GS can contribute to a higher diagnostic yield and a better understanding of the genetic landscape of the lissencephaly spectrum.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
