Dinah Clark, Robert Burns, Michelle S. Bloom, Karen Phaik Har Lim, Lili Li, Lisa M. Vincent, Jing Xie, Yuan Xue, Sumit Punj
{"title":"Heterozygous loss of function variants in IFT140 are associated with polycystic kidney disease","authors":"Dinah Clark, Robert Burns, Michelle S. Bloom, Karen Phaik Har Lim, Lili Li, Lisa M. Vincent, Jing Xie, Yuan Xue, Sumit Punj","doi":"10.1002/ajmg.a.63841","DOIUrl":null,"url":null,"abstract":"<p>Autosomal dominant polycystic kidney disease (ADPKD) affects 1 in 1000 adults. Most cases result from causative <i>PKD1</i> or <i>PKD2</i> variants. <i>HNF1B, GANAB</i> and <i>ALG9</i> variants are also associated with ADPKD. Recent evidence indicates that monoallelic loss-of-function (LoF) <i>IFT140</i> variants are a cause for non-syndromic ADPKD. We describe 368 patients with <i>IFT140</i> LoF variants and a spectrum of phenotypic findings that support the association of <i>IFT140</i> with PKD. We reviewed patients with an unknown cause for their cystic disease and those with heterozygous LoF <i>IFT140</i> variants classified as pathogenic or likely pathogenic from a cohort that received genetic testing using a panel of 385 renal disease-associated genes. <i>IFT140</i> LoF variants were significantly enriched in patients with cystic disease when compared with those without cystic disease. A cystic phenotype was reported in 223 of the 368 (60.6%) individuals harboring an <i>IFT140</i> LoF variant, 98% of which had no other identified cause for their cystic disease. Of 122 unique LoF <i>IFT140</i> variants identified, 56 (46%) were frameshift, 38 (31%) nonsense, 22 (18%) splice site and 6 (5%) exon-level deletions. Only six <i>IFT140</i> individuals were reported with end-stage kidney disease, consistent with observed milder clinical presentations in <i>IFT140-</i>related PKD. This study offers further evidence for the involvement of LoF <i>IFT140</i> variants in PKD, particularly when no additional molecular etiology has been identified.</p>","PeriodicalId":7507,"journal":{"name":"American Journal of Medical Genetics Part A","volume":"194 12","pages":""},"PeriodicalIF":1.7000,"publicationDate":"2024-08-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ajmg.a.63841","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"American Journal of Medical Genetics Part A","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63841","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
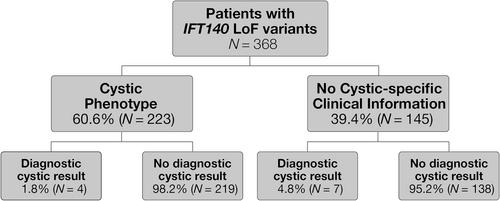
Autosomal dominant polycystic kidney disease (ADPKD) affects 1 in 1000 adults. Most cases result from causative PKD1 or PKD2 variants. HNF1B, GANAB and ALG9 variants are also associated with ADPKD. Recent evidence indicates that monoallelic loss-of-function (LoF) IFT140 variants are a cause for non-syndromic ADPKD. We describe 368 patients with IFT140 LoF variants and a spectrum of phenotypic findings that support the association of IFT140 with PKD. We reviewed patients with an unknown cause for their cystic disease and those with heterozygous LoF IFT140 variants classified as pathogenic or likely pathogenic from a cohort that received genetic testing using a panel of 385 renal disease-associated genes. IFT140 LoF variants were significantly enriched in patients with cystic disease when compared with those without cystic disease. A cystic phenotype was reported in 223 of the 368 (60.6%) individuals harboring an IFT140 LoF variant, 98% of which had no other identified cause for their cystic disease. Of 122 unique LoF IFT140 variants identified, 56 (46%) were frameshift, 38 (31%) nonsense, 22 (18%) splice site and 6 (5%) exon-level deletions. Only six IFT140 individuals were reported with end-stage kidney disease, consistent with observed milder clinical presentations in IFT140-related PKD. This study offers further evidence for the involvement of LoF IFT140 variants in PKD, particularly when no additional molecular etiology has been identified.
期刊介绍:
The American Journal of Medical Genetics - Part A (AJMG) gives you continuous coverage of all biological and medical aspects of genetic disorders and birth defects, as well as in-depth documentation of phenotype analysis within the current context of genotype/phenotype correlations. In addition to Part A , AJMG also publishes two other parts:
Part B: Neuropsychiatric Genetics , covering experimental and clinical investigations of the genetic mechanisms underlying neurologic and psychiatric disorders.
Part C: Seminars in Medical Genetics , guest-edited collections of thematic reviews of topical interest to the readership of AJMG .


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
