Sara Lettieri, Francesco Bonella, Vincenzo Alfredo Marando, Alessandro N Franciosi, Angelo Guido Corsico, Ilaria Campo
{"title":"Pathogenesis-driven treatment of primary pulmonary alveolar proteinosis.","authors":"Sara Lettieri, Francesco Bonella, Vincenzo Alfredo Marando, Alessandro N Franciosi, Angelo Guido Corsico, Ilaria Campo","doi":"10.1183/16000617.0064-2024","DOIUrl":null,"url":null,"abstract":"<p><p>Pulmonary alveolar proteinosis (PAP) is a syndrome that results from the accumulation of lipoproteinaceous material in the alveolar space. According to the underlying pathogenetic mechanisms, three different forms have been identified, namely primary, secondary and congenital. Primary PAP is caused by disruption of granulocyte-macrophage colony-stimulating factor (GM-CSF) signalling due to the presence of neutralising autoantibodies (autoimmune PAP) or GM-CSF receptor genetic defects (hereditary PAP), which results in dysfunctional alveolar macrophages with reduced phagocytic clearance of particles, cholesterol and surfactant. The serum level of GM-CSF autoantibody is the only disease-specific biomarker of autoimmune PAP, although it does not correlate with disease severity. In PAP patients with normal serum GM-CSF autoantibody levels, elevated serum GM-CSF levels is highly suspicious for hereditary PAP. Several biomarkers have been correlated with disease severity, although they are not specific for PAP. These include lactate dehydrogenase, cytokeratin 19 fragment 21.1, carcinoembryonic antigen, neuron-specific enolase, surfactant proteins, Krebs von Lungen 6, chitinase-3-like protein 1 and monocyte chemotactic proteins. Finally, increased awareness of the disease mechanisms has led to the development of pathogenesis-based treatments, such as GM-CSF augmentation and cholesterol-targeting therapies.</p>","PeriodicalId":12166,"journal":{"name":"European Respiratory Review","volume":"33 173","pages":""},"PeriodicalIF":10.4000,"publicationDate":"2024-08-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11322829/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Respiratory Review","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1183/16000617.0064-2024","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/1 0:00:00","PubModel":"Print","JCR":"Q1","JCRName":"RESPIRATORY SYSTEM","Score":null,"Total":0}
引用次数: 0
Abstract
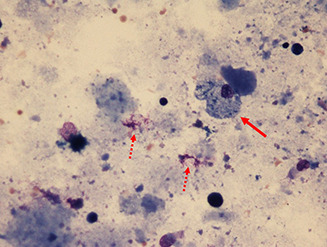
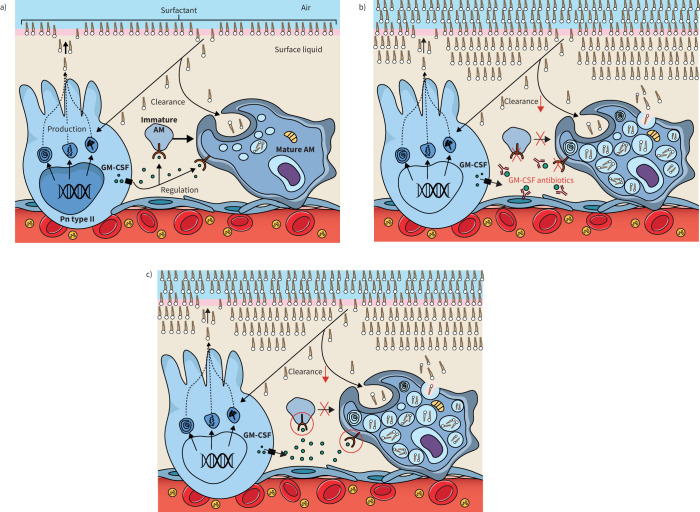
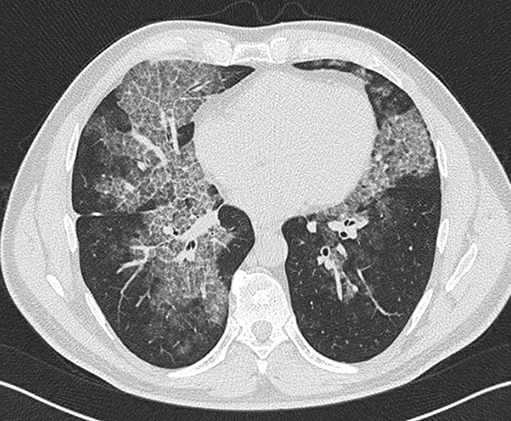
Pulmonary alveolar proteinosis (PAP) is a syndrome that results from the accumulation of lipoproteinaceous material in the alveolar space. According to the underlying pathogenetic mechanisms, three different forms have been identified, namely primary, secondary and congenital. Primary PAP is caused by disruption of granulocyte-macrophage colony-stimulating factor (GM-CSF) signalling due to the presence of neutralising autoantibodies (autoimmune PAP) or GM-CSF receptor genetic defects (hereditary PAP), which results in dysfunctional alveolar macrophages with reduced phagocytic clearance of particles, cholesterol and surfactant. The serum level of GM-CSF autoantibody is the only disease-specific biomarker of autoimmune PAP, although it does not correlate with disease severity. In PAP patients with normal serum GM-CSF autoantibody levels, elevated serum GM-CSF levels is highly suspicious for hereditary PAP. Several biomarkers have been correlated with disease severity, although they are not specific for PAP. These include lactate dehydrogenase, cytokeratin 19 fragment 21.1, carcinoembryonic antigen, neuron-specific enolase, surfactant proteins, Krebs von Lungen 6, chitinase-3-like protein 1 and monocyte chemotactic proteins. Finally, increased awareness of the disease mechanisms has led to the development of pathogenesis-based treatments, such as GM-CSF augmentation and cholesterol-targeting therapies.
期刊介绍:
The European Respiratory Review (ERR) is an open-access journal published by the European Respiratory Society (ERS), serving as a vital resource for respiratory professionals by delivering updates on medicine, science, and surgery in the field. ERR features state-of-the-art review articles, editorials, correspondence, and summaries of recent research findings and studies covering a wide range of topics including COPD, asthma, pulmonary hypertension, interstitial lung disease, lung cancer, tuberculosis, and pulmonary infections. Articles are published continuously and compiled into quarterly issues within a single annual volume.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
