Hannah Lahey, Haewon Shin, Katherine Myers, Kim L. McBride
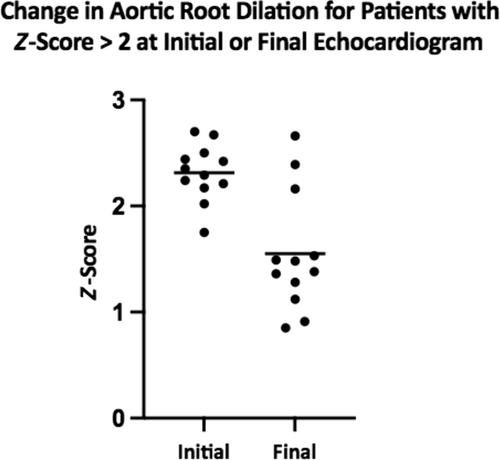
{"title":"Longitudinal echocardiography in pediatric patients with hypermobile Ehlers-Danlos syndrome","authors":"Hannah Lahey, Haewon Shin, Katherine Myers, Kim L. McBride","doi":"10.1002/ajmg.a.63844","DOIUrl":null,"url":null,"abstract":"<p>Vascular Ehlers-Danlos, Marfan and Loeys-Dietz syndromes have increased risk of aortic dilation and dissection. Previous early studies showed hypermobile Ehlers-Danlos syndrome (hEDS) may also have increased risk, with echocardiography screening recommended; subsequent studies have not confirmed the risk or recommended echocardiography. This pediatric-based study assessed aortic dilation prevalence in those with hEDS by serial echocardiographic examinations and assessed family history for aortic dissections. We retrospectively identified individuals with hEDS who had echocardiography studies from the electronic medical records at one pediatric center. Aortic root <i>Z</i>-scores >2.0 were found in 15/225 subjects (average age 12.9 years) on initial echocardiograms, with no <i>Z</i>-score >3.0. Subsequent studies (<i>n</i> = 68) found statistically significant decline in aortic root <i>Z</i>-scores. Repeat echocardiography in those with initial aortic root <i>Z</i>-score >2.0 (<i>n</i> = 10) demonstrated a decline in <i>Z</i> score <2.0 in seven. On final examination, 9/225 (4.0%) had a <i>Z</i>-score >2.0, not statistically different from the general population. No aortic dissection occurred in first- or second-degree relatives. In conclusion, aortic root dilation rate in hEDS is likely not different from the general population. We propose that in the absence of other cardiac findings or suspicion for another disorder, echocardiography is not required in hEDS.</p>","PeriodicalId":7507,"journal":{"name":"American Journal of Medical Genetics Part A","volume":"194 12","pages":""},"PeriodicalIF":1.7000,"publicationDate":"2024-08-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/ajmg.a.63844","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"American Journal of Medical Genetics Part A","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.63844","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Vascular Ehlers-Danlos, Marfan and Loeys-Dietz syndromes have increased risk of aortic dilation and dissection. Previous early studies showed hypermobile Ehlers-Danlos syndrome (hEDS) may also have increased risk, with echocardiography screening recommended; subsequent studies have not confirmed the risk or recommended echocardiography. This pediatric-based study assessed aortic dilation prevalence in those with hEDS by serial echocardiographic examinations and assessed family history for aortic dissections. We retrospectively identified individuals with hEDS who had echocardiography studies from the electronic medical records at one pediatric center. Aortic root Z-scores >2.0 were found in 15/225 subjects (average age 12.9 years) on initial echocardiograms, with no Z-score >3.0. Subsequent studies (n = 68) found statistically significant decline in aortic root Z-scores. Repeat echocardiography in those with initial aortic root Z-score >2.0 (n = 10) demonstrated a decline in Z score <2.0 in seven. On final examination, 9/225 (4.0%) had a Z-score >2.0, not statistically different from the general population. No aortic dissection occurred in first- or second-degree relatives. In conclusion, aortic root dilation rate in hEDS is likely not different from the general population. We propose that in the absence of other cardiac findings or suspicion for another disorder, echocardiography is not required in hEDS.
期刊介绍:
The American Journal of Medical Genetics - Part A (AJMG) gives you continuous coverage of all biological and medical aspects of genetic disorders and birth defects, as well as in-depth documentation of phenotype analysis within the current context of genotype/phenotype correlations. In addition to Part A , AJMG also publishes two other parts:
Part B: Neuropsychiatric Genetics , covering experimental and clinical investigations of the genetic mechanisms underlying neurologic and psychiatric disorders.
Part C: Seminars in Medical Genetics , guest-edited collections of thematic reviews of topical interest to the readership of AJMG .


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
