Arezoo Fallah, Seyed Asghar Havaei, Hamid Sedighian, Reza Kachuei and Abbas Ali Imani Fooladi
{"title":"Prediction of aptamer affinity using an artificial intelligence approach","authors":"Arezoo Fallah, Seyed Asghar Havaei, Hamid Sedighian, Reza Kachuei and Abbas Ali Imani Fooladi","doi":"10.1039/D4TB00909F","DOIUrl":null,"url":null,"abstract":"<p >Aptamers are oligonucleotide sequences that can connect to particular target molecules, similar to monoclonal antibodies. They can be chosen by systematic evolution of ligands by exponential enrichment (SELEX), and are modifiable and can be synthesized. Even if the SELEX approach has been improved a lot, it is frequently challenging and time-consuming to identify aptamers experimentally. In particular, structure-based methods are the most used in computer-aided design and development of aptamers. For this purpose, numerous web-based platforms have been suggested for the purpose of forecasting the secondary structure and 3D configurations of RNAs and DNAs. Also, molecular docking and molecular dynamics (MD), which are commonly utilized in protein compound selection by structural information, are suitable for aptamer selection. On the other hand, from a large number of sequences, artificial intelligence (AI) may be able to quickly discover the possible aptamer candidates. Conversely, sophisticated machine and deep-learning (DL) models have demonstrated efficacy in forecasting the binding properties between ligands and targets during drug discovery; as such, they may provide a reliable and precise method for forecasting the binding of aptamers to targets. This research looks at advancements in AI pipelines and strategies for aptamer binding ability prediction, such as machine and deep learning, as well as structure-based approaches, molecular dynamics and molecular docking simulation methods.</p>","PeriodicalId":83,"journal":{"name":"Journal of Materials Chemistry B","volume":" 36","pages":" 8825-8842"},"PeriodicalIF":6.1000,"publicationDate":"2024-08-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Materials Chemistry B","FirstCategoryId":"1","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/tb/d4tb00909f","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATERIALS SCIENCE, BIOMATERIALS","Score":null,"Total":0}
引用次数: 0
Abstract
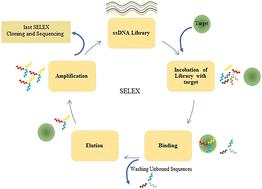
Aptamers are oligonucleotide sequences that can connect to particular target molecules, similar to monoclonal antibodies. They can be chosen by systematic evolution of ligands by exponential enrichment (SELEX), and are modifiable and can be synthesized. Even if the SELEX approach has been improved a lot, it is frequently challenging and time-consuming to identify aptamers experimentally. In particular, structure-based methods are the most used in computer-aided design and development of aptamers. For this purpose, numerous web-based platforms have been suggested for the purpose of forecasting the secondary structure and 3D configurations of RNAs and DNAs. Also, molecular docking and molecular dynamics (MD), which are commonly utilized in protein compound selection by structural information, are suitable for aptamer selection. On the other hand, from a large number of sequences, artificial intelligence (AI) may be able to quickly discover the possible aptamer candidates. Conversely, sophisticated machine and deep-learning (DL) models have demonstrated efficacy in forecasting the binding properties between ligands and targets during drug discovery; as such, they may provide a reliable and precise method for forecasting the binding of aptamers to targets. This research looks at advancements in AI pipelines and strategies for aptamer binding ability prediction, such as machine and deep learning, as well as structure-based approaches, molecular dynamics and molecular docking simulation methods.
期刊介绍:
Journal of Materials Chemistry A, B & C cover high quality studies across all fields of materials chemistry. The journals focus on those theoretical or experimental studies that report new understanding, applications, properties and synthesis of materials. Journal of Materials Chemistry A, B & C are separated by the intended application of the material studied. Broadly, applications in energy and sustainability are of interest to Journal of Materials Chemistry A, applications in biology and medicine are of interest to Journal of Materials Chemistry B, and applications in optical, magnetic and electronic devices are of interest to Journal of Materials Chemistry C.Journal of Materials Chemistry B is a Transformative Journal and Plan S compliant. Example topic areas within the scope of Journal of Materials Chemistry B are listed below. This list is neither exhaustive nor exclusive:
Antifouling coatings
Biocompatible materials
Bioelectronics
Bioimaging
Biomimetics
Biomineralisation
Bionics
Biosensors
Diagnostics
Drug delivery
Gene delivery
Immunobiology
Nanomedicine
Regenerative medicine & Tissue engineering
Scaffolds
Soft robotics
Stem cells
Therapeutic devices




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
