Freddie L. Braddock, Jessica C. Gardner, Nihar Bhattacharyya, Beatriz Sanchez-Pintado, Marcos Costa, Christina Zarouchlioti, Anita Szabo, Petra Lišková, Michael E. Cheetham, Robert D. Young, Caroline Thaung, Alice E. Davidson, Stephen J. Tuft, Alison J. Hardcastle
{"title":"Autosomal dominant stromal corneal dystrophy associated with a SPARCL1 missense variant","authors":"Freddie L. Braddock, Jessica C. Gardner, Nihar Bhattacharyya, Beatriz Sanchez-Pintado, Marcos Costa, Christina Zarouchlioti, Anita Szabo, Petra Lišková, Michael E. Cheetham, Robert D. Young, Caroline Thaung, Alice E. Davidson, Stephen J. Tuft, Alison J. Hardcastle","doi":"10.1038/s41431-024-01687-8","DOIUrl":null,"url":null,"abstract":"Corneal dystrophies are phenotypically and genetically heterogeneous, often resulting in visual impairment caused by corneal opacification. We investigated the genetic cause of an autosomal dominant corneal stromal dystrophy in a pedigree with eight affected individuals in three generations. Affected individuals had diffuse central stromal opacity, with reduced visual acuity in older family members. Histopathology of affected cornea tissue removed during surgery revealed mild stromal textural alterations with alcianophilic deposits. Whole genome sequence data were generated for four affected individuals. No rare variants (MAF < 0.001) were identified in established corneal dystrophy genes. However, a novel heterozygous missense variant in exon 4 of SPARCL1, NM_004684: c.334G > A; p.(Glu112Lys), which is predicted to be damaging, segregated with disease. SPARC-like protein 1 (SPARCL1) is a secreted matricellular protein involved in cell migration, cell adhesion, tissue repair, and remodelling. Interestingly, SPARCL1 has been shown to regulate decorin. Heterozygous variants in DCN, encoding decorin, cause autosomal dominant congenital stromal corneal dystrophy, suggesting a common pathogenic pathway. Therefore, we performed immunohistochemistry to compare SPARCL1 and decorin localisation in corneal tissue from an affected family member and an unaffected control. Strikingly, the level of decorin was significantly decreased in the corneal stroma of the affected tissue, and SPARCL1 appeared to be retained in the epithelium. In summary, we describe a novel autosomal dominant corneal stromal dystrophy associated with a missense variant in SPARCL1, extending the phenotypic and genetic heterogeneity of inherited corneal disease.","PeriodicalId":12016,"journal":{"name":"European Journal of Human Genetics","volume":"32 12","pages":"1583-1589"},"PeriodicalIF":4.6000,"publicationDate":"2024-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s41431-024-01687-8.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s41431-024-01687-8","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
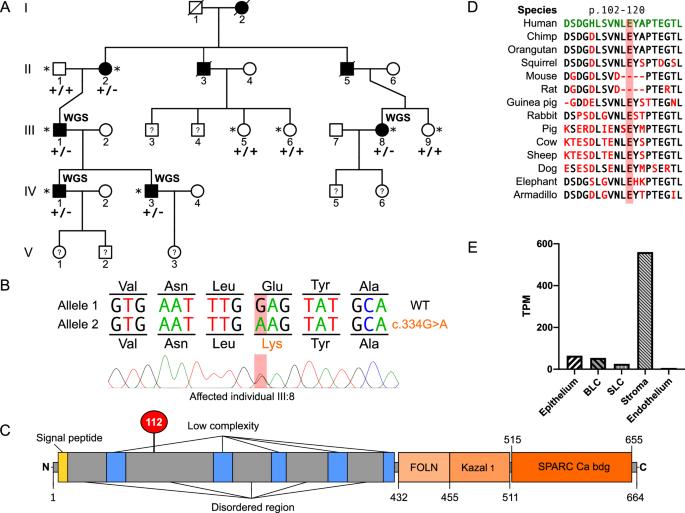
Corneal dystrophies are phenotypically and genetically heterogeneous, often resulting in visual impairment caused by corneal opacification. We investigated the genetic cause of an autosomal dominant corneal stromal dystrophy in a pedigree with eight affected individuals in three generations. Affected individuals had diffuse central stromal opacity, with reduced visual acuity in older family members. Histopathology of affected cornea tissue removed during surgery revealed mild stromal textural alterations with alcianophilic deposits. Whole genome sequence data were generated for four affected individuals. No rare variants (MAF < 0.001) were identified in established corneal dystrophy genes. However, a novel heterozygous missense variant in exon 4 of SPARCL1, NM_004684: c.334G > A; p.(Glu112Lys), which is predicted to be damaging, segregated with disease. SPARC-like protein 1 (SPARCL1) is a secreted matricellular protein involved in cell migration, cell adhesion, tissue repair, and remodelling. Interestingly, SPARCL1 has been shown to regulate decorin. Heterozygous variants in DCN, encoding decorin, cause autosomal dominant congenital stromal corneal dystrophy, suggesting a common pathogenic pathway. Therefore, we performed immunohistochemistry to compare SPARCL1 and decorin localisation in corneal tissue from an affected family member and an unaffected control. Strikingly, the level of decorin was significantly decreased in the corneal stroma of the affected tissue, and SPARCL1 appeared to be retained in the epithelium. In summary, we describe a novel autosomal dominant corneal stromal dystrophy associated with a missense variant in SPARCL1, extending the phenotypic and genetic heterogeneity of inherited corneal disease.
期刊介绍:
The European Journal of Human Genetics is the official journal of the European Society of Human Genetics, publishing high-quality, original research papers, short reports and reviews in the rapidly expanding field of human genetics and genomics. It covers molecular, clinical and cytogenetics, interfacing between advanced biomedical research and the clinician, and bridging the great diversity of facilities, resources and viewpoints in the genetics community.
Key areas include:
-Monogenic and multifactorial disorders
-Development and malformation
-Hereditary cancer
-Medical Genomics
-Gene mapping and functional studies
-Genotype-phenotype correlations
-Genetic variation and genome diversity
-Statistical and computational genetics
-Bioinformatics
-Advances in diagnostics
-Therapy and prevention
-Animal models
-Genetic services
-Community genetics



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
