{"title":"Molecular simulations of understanding the Zn2+ ion structure, dynamics and thermodynamic properties in water in ionic liquids","authors":"Raunak Katiyar, Praveenkumar Sappidi","doi":"10.1016/j.chemphys.2024.112424","DOIUrl":null,"url":null,"abstract":"<div><p>We utilize all-atom molecular dynamics simulations to explore the intermolecular structure, dynamics and thermodynamic properties of Zn<sup>2+</sup> ions in water-in-ionic liquid. Two ionic liquid systems featuring the same cation 1-ethyl-3-methyl-imidazolium [EMIM]<sup>+</sup> and distinct anions tetrafluoroborate [BF<sub>4</sub>]<sup>−</sup> and hexafluorophosphate [PF<sub>6</sub>]<sup>−</sup> are considered. We consider the water (H<sub>2</sub>O) mole fractions (x) varying from 0.33 to 0.71. We observe a significant interactions of Zn<sup>2+</sup> ions with water in the case of [BF<sub>4</sub>]<sup>−</sup> when compared to [PF<sub>6</sub>]<sup>−</sup>. On the other hand Zn<sup>2+</sup> ions mobility rises more in [EMIM]<sup>+</sup>[BF<sub>4</sub>]<sup>−</sup> as compared to [EMIM]<sup>+</sup>[PF<sub>6</sub>]<sup>−</sup> with x. Higher self-diffusion (D) of Zn<sup>2+</sup> ions is seen in the case of [EMIM]<sup>+</sup>[BF<sub>4</sub>]<sup>−</sup>. The ionic conductivity (σ) of [EMIM]<sup>+</sup>[BF<sub>4</sub>]<sup>−</sup> is greater compared to [EMIM]<sup>+</sup>[PF<sub>6</sub>]<sup>−</sup> with the rise in x. Overall, this article furnishes in-depth molecular-level insights into the behaviour of Zn<sup>2+</sup> ions in the presence of water mixed ionic liquid electrolytes.</p></div>","PeriodicalId":272,"journal":{"name":"Chemical Physics","volume":"587 ","pages":"Article 112424"},"PeriodicalIF":2.4000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0301010424002532","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/15 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
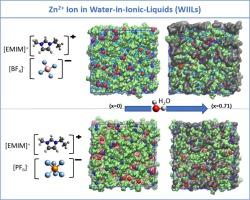
Abstract
We utilize all-atom molecular dynamics simulations to explore the intermolecular structure, dynamics and thermodynamic properties of Zn2+ ions in water-in-ionic liquid. Two ionic liquid systems featuring the same cation 1-ethyl-3-methyl-imidazolium [EMIM]+ and distinct anions tetrafluoroborate [BF4]− and hexafluorophosphate [PF6]− are considered. We consider the water (H2O) mole fractions (x) varying from 0.33 to 0.71. We observe a significant interactions of Zn2+ ions with water in the case of [BF4]− when compared to [PF6]−. On the other hand Zn2+ ions mobility rises more in [EMIM]+[BF4]− as compared to [EMIM]+[PF6]− with x. Higher self-diffusion (D) of Zn2+ ions is seen in the case of [EMIM]+[BF4]−. The ionic conductivity (σ) of [EMIM]+[BF4]− is greater compared to [EMIM]+[PF6]− with the rise in x. Overall, this article furnishes in-depth molecular-level insights into the behaviour of Zn2+ ions in the presence of water mixed ionic liquid electrolytes.
期刊介绍:
Chemical Physics publishes experimental and theoretical papers on all aspects of chemical physics. In this journal, experiments are related to theory, and in turn theoretical papers are related to present or future experiments. Subjects covered include: spectroscopy and molecular structure, interacting systems, relaxation phenomena, biological systems, materials, fundamental problems in molecular reactivity, molecular quantum theory and statistical mechanics. Computational chemistry studies of routine character are not appropriate for this journal.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
