Design, synthesis and biological evaluation of thieno[3,2-c]pyrazol-urea derivatives as potent glycogen synthase kinase 3β inhibitors based on the DFG-out conformation
Ning Yan , Hong-Yan Liu , Ting-Ting Kong , Zi-Hao Kong , Ling-Yun Li , Xin Ma , Yan-Li Zeng , Mei-Jun Wang , Long-Qian Tang , Cheng-Mei Zhang , Zhao-Peng Liu , Chao Liu
{"title":"Design, synthesis and biological evaluation of thieno[3,2-c]pyrazol-urea derivatives as potent glycogen synthase kinase 3β inhibitors based on the DFG-out conformation","authors":"Ning Yan , Hong-Yan Liu , Ting-Ting Kong , Zi-Hao Kong , Ling-Yun Li , Xin Ma , Yan-Li Zeng , Mei-Jun Wang , Long-Qian Tang , Cheng-Mei Zhang , Zhao-Peng Liu , Chao Liu","doi":"10.1016/j.bmcl.2024.129932","DOIUrl":null,"url":null,"abstract":"<div><p>Glycogen synthase kinase 3β (GSK-3β) is a potential therapeutic target for the treatment of a variety of human diseases. Here, we report the design and synthesis of a series of thieno[3,2-<em>c</em>]pyrazol-urea derivatives and evaluation of their GSK-3β inhibitory activity. Among these analogues, the compound without substitution on terminal phenyl ring (<strong>3a</strong>) was found to be the most potent GSK-3β inhibitor with an IC<sub>50</sub> of 74.4 nM, while substitution on the terminal phenyl (<strong>3b</strong>–<strong>3p</strong>) led to decreased potency, independent of the position, size, or electronic properties of the substituents. Kinase selectivity assay revealed that <strong>3a</strong> showed good selectivity over a panel of kinases, but was less selective over CDK1, CDK2 and CDK5. Additionally, the pharmacological properties of the synthesized compounds were investigated computationally by the SwissADME and the results showed that most of the compounds have good ADME profiles.</p></div>","PeriodicalId":256,"journal":{"name":"Bioorganic & Medicinal Chemistry Letters","volume":"112 ","pages":"Article 129932"},"PeriodicalIF":2.2000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioorganic & Medicinal Chemistry Letters","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0960894X24003342","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/28 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
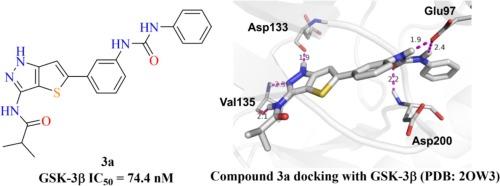
Glycogen synthase kinase 3β (GSK-3β) is a potential therapeutic target for the treatment of a variety of human diseases. Here, we report the design and synthesis of a series of thieno[3,2-c]pyrazol-urea derivatives and evaluation of their GSK-3β inhibitory activity. Among these analogues, the compound without substitution on terminal phenyl ring (3a) was found to be the most potent GSK-3β inhibitor with an IC50 of 74.4 nM, while substitution on the terminal phenyl (3b–3p) led to decreased potency, independent of the position, size, or electronic properties of the substituents. Kinase selectivity assay revealed that 3a showed good selectivity over a panel of kinases, but was less selective over CDK1, CDK2 and CDK5. Additionally, the pharmacological properties of the synthesized compounds were investigated computationally by the SwissADME and the results showed that most of the compounds have good ADME profiles.
期刊介绍:
Bioorganic & Medicinal Chemistry Letters presents preliminary experimental or theoretical research results of outstanding significance and timeliness on all aspects of science at the interface of chemistry and biology and on major advances in drug design and development. The journal publishes articles in the form of communications reporting experimental or theoretical results of special interest, and strives to provide maximum dissemination to a large, international audience.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
