{"title":"Efficient Shift-and-Invert Preconditioning for Multi-GPU Accelerated Density Functional Calculations.","authors":"Jeheon Woo, Woo Youn Kim, Sunghwan Choi","doi":"10.1021/acs.jctc.4c00721","DOIUrl":null,"url":null,"abstract":"<p><p>To accelerate the iterative diagonalization of electronic structure calculations, we propose a new inexact shift-and-invert (ISI) preconditioning method. The key idea is to improve shift values in the ISI preconditioning to be closer to the exact eigenvalues, leading to a significant boost in the convergence speed of the iterative diagonalization. Furthermore, we adopted a preconditioned conjugate gradient solver to rapidly evaluate an inversion process. Finally, we accelerated overall processes, including the proposed modification, with state-of-the-art graphical processing units (GPUs) and assessed its parallel efficiency with real-space density functional calculations of 1D, 2D, and 3D periodic systems. Our method attains both fast diagonalization convergence and high multi-GPU parallel efficiency. This is evident from the fact that single-point density functional calculations for hundreds of atom systems can be done in approximately 10 s using 8 GPUs. The proposed method can be generally applied to any electronic structure calculation methods involving large-scale diagonalizations.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"7443-7452"},"PeriodicalIF":5.5000,"publicationDate":"2024-09-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11391578/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00721","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/27 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
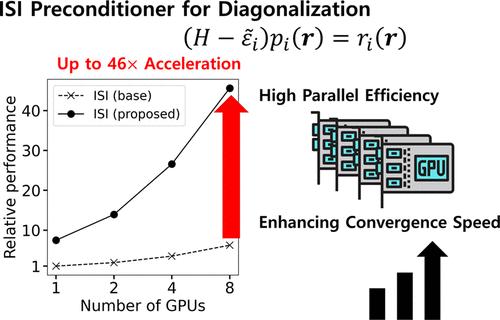
To accelerate the iterative diagonalization of electronic structure calculations, we propose a new inexact shift-and-invert (ISI) preconditioning method. The key idea is to improve shift values in the ISI preconditioning to be closer to the exact eigenvalues, leading to a significant boost in the convergence speed of the iterative diagonalization. Furthermore, we adopted a preconditioned conjugate gradient solver to rapidly evaluate an inversion process. Finally, we accelerated overall processes, including the proposed modification, with state-of-the-art graphical processing units (GPUs) and assessed its parallel efficiency with real-space density functional calculations of 1D, 2D, and 3D periodic systems. Our method attains both fast diagonalization convergence and high multi-GPU parallel efficiency. This is evident from the fact that single-point density functional calculations for hundreds of atom systems can be done in approximately 10 s using 8 GPUs. The proposed method can be generally applied to any electronic structure calculation methods involving large-scale diagonalizations.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
