{"title":"Treatment-resistant schizophrenia with 22q11.2 deletion and additional genetic defects.","authors":"Sawako Furukawa, Shusei Arafuka, Hidekazu Kato, Tomoo Ogi, Norio Ozaki, Masashi Ikeda, Itaru Kushima","doi":"10.1002/npr2.12477","DOIUrl":null,"url":null,"abstract":"<p><p>We report a case of a 61-year-old female with 22q11.2 deletion syndrome (22q11.2DS) and a novel heterozygous nonsense variant in MAP1A, identified through whole-genome sequencing (WGS). The patient presented with intellectual developmental disorder, treatment-resistant schizophrenia (SCZ), and multiple congenital anomalies. Despite aggressive pharmacotherapy, she experienced persistent auditory hallucinations and negative symptoms. WGS revealed a 3 Mb deletion at 22q11.2 and a nonsense variant in MAP1A (c.4652T>G, p.Leu1551*). MAP1A, encoding microtubule-associated protein 1A, is crucial for axon and dendrite development and has been implicated in autism spectrum disorder and SCZ. The MAP1A variant may contribute to the severe psychiatric phenotype, as it is thought to influence synaptic plasticity, a process also affected by 22q11.2 deletion. This case highlights the importance of WGS in identifying additional pathogenic variants that may explain phenotypic variability in 22q11.2DS. Thus, WGS can lead to a better understanding of the genetic architecture of 22q11.2DS. However, further studies are needed to elucidate the role of secondary genetic contributors in the diverse clinical presentations of 22q11.2DS.</p>","PeriodicalId":19137,"journal":{"name":"Neuropsychopharmacology Reports","volume":" ","pages":"847-851"},"PeriodicalIF":2.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11609749/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neuropsychopharmacology Reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1002/npr2.12477","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/27 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
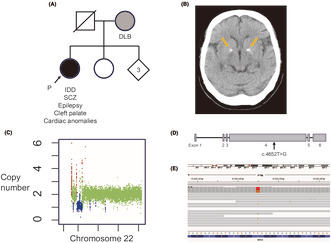
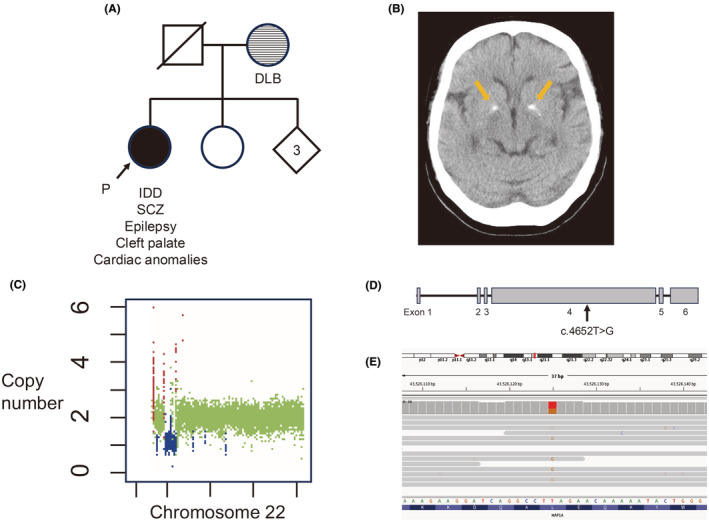
We report a case of a 61-year-old female with 22q11.2 deletion syndrome (22q11.2DS) and a novel heterozygous nonsense variant in MAP1A, identified through whole-genome sequencing (WGS). The patient presented with intellectual developmental disorder, treatment-resistant schizophrenia (SCZ), and multiple congenital anomalies. Despite aggressive pharmacotherapy, she experienced persistent auditory hallucinations and negative symptoms. WGS revealed a 3 Mb deletion at 22q11.2 and a nonsense variant in MAP1A (c.4652T>G, p.Leu1551*). MAP1A, encoding microtubule-associated protein 1A, is crucial for axon and dendrite development and has been implicated in autism spectrum disorder and SCZ. The MAP1A variant may contribute to the severe psychiatric phenotype, as it is thought to influence synaptic plasticity, a process also affected by 22q11.2 deletion. This case highlights the importance of WGS in identifying additional pathogenic variants that may explain phenotypic variability in 22q11.2DS. Thus, WGS can lead to a better understanding of the genetic architecture of 22q11.2DS. However, further studies are needed to elucidate the role of secondary genetic contributors in the diverse clinical presentations of 22q11.2DS.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
