Bi Huang, Cong Fan, Ken Chen, Jiahua Rao, Peihua Ou, Chong Tian, Yuedong Yang, David N Cooper, Huiying Zhao
{"title":"VCAT: an integrated variant function annotation tools.","authors":"Bi Huang, Cong Fan, Ken Chen, Jiahua Rao, Peihua Ou, Chong Tian, Yuedong Yang, David N Cooper, Huiying Zhao","doi":"10.1007/s00439-024-02699-6","DOIUrl":null,"url":null,"abstract":"<p><p>The development of sequencing technology has promoted discovery of variants in the human genome. Identifying functions of these variants is important for us to link genotype to phenotype, and to diagnose diseases. However, it usually requires researchers to visit multiple databases. Here, we presented a one-stop webserver for variant function annotation tools (VCAT, https://biomed.nscc-gz.cn/zhaolab/VCAT/ ) that is the first one connecting variant to functions via the epigenome, protein, drug and RNA. VCAT is also the first one to make all annotations visualized in interactive charts or molecular structures. VCAT allows users to upload data in VCF format, and download results via a URL. Moreover, VCAT has annotated a huge number (1,262,041,068) of variants collected from dbSNP, 1000 Genomes projects, gnomAD, ICGC, TCGA, and HPRC Pangenome project. For these variants, users are able to searcher their functions, related diseases and drugs from VCAT. In summary, VCAT provides a one-stop webserver to explore the potential functions of human genomic variants including their relationship with diseases and drugs.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":" ","pages":"1311-1322"},"PeriodicalIF":3.6000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-024-02699-6","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/27 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
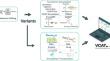
The development of sequencing technology has promoted discovery of variants in the human genome. Identifying functions of these variants is important for us to link genotype to phenotype, and to diagnose diseases. However, it usually requires researchers to visit multiple databases. Here, we presented a one-stop webserver for variant function annotation tools (VCAT, https://biomed.nscc-gz.cn/zhaolab/VCAT/ ) that is the first one connecting variant to functions via the epigenome, protein, drug and RNA. VCAT is also the first one to make all annotations visualized in interactive charts or molecular structures. VCAT allows users to upload data in VCF format, and download results via a URL. Moreover, VCAT has annotated a huge number (1,262,041,068) of variants collected from dbSNP, 1000 Genomes projects, gnomAD, ICGC, TCGA, and HPRC Pangenome project. For these variants, users are able to searcher their functions, related diseases and drugs from VCAT. In summary, VCAT provides a one-stop webserver to explore the potential functions of human genomic variants including their relationship with diseases and drugs.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
