Yutaka Hasegawa, Toshie Segawa, Ai Chida, Eriko Yoshida, Hirofumi Kinno, Hiraku Chiba, Tomoyasu Oda, Yoshihiko Takahashi, Koji Nata, Yasushi Ishigaki
{"title":"A novel frameshift variant of GATA3 (p.Ala17ProfsTer178) responsible for HDR syndrome in a Japanese family.","authors":"Yutaka Hasegawa, Toshie Segawa, Ai Chida, Eriko Yoshida, Hirofumi Kinno, Hiraku Chiba, Tomoyasu Oda, Yoshihiko Takahashi, Koji Nata, Yasushi Ishigaki","doi":"10.1507/endocrj.EJ24-0147","DOIUrl":null,"url":null,"abstract":"<p><p>HDR syndrome is an autosomal dominant disorder characterized by hypoparathyroidism (H), deafness (D), and renal dysplasia (R) caused by genetic variants of the GATA3 gene. We present the case of a 38-year-old Japanese man with HDR syndrome who exhibited hypoparathyroidism, sensorineural deafness, renal dysfunction, severe symptomatic hypocalcemia with Chvostek's and Trousseau's signs, and QT prolongation on electrocardiography. He had a family history of deafness and hypocalcemia. Genetic testing revealed a novel GATA3 gene variant at exon 2 (c.48delC), which induces a frameshift resulting in termination at codon 178, causing HDR syndrome. We summarized 45 Japanese cases of HDR syndrome with regard to the mode of onset (familial or sporadic) and the age at diagnosis. In addition, we summarized all previous cases of HDR syndrome with GATA3 gene variants. Mapping of previously reported genetic variants in HDR syndrome revealed that most missense variants were observed at exons 4 and 5 regions in the GATA3 gene. These two regions contain zinc finger domains, demonstrating their functional importance in GATA3 transcription. This review of literature provides a useful reference for diagnosing HDR syndrome and predicting the related future manifestations.</p>","PeriodicalId":11631,"journal":{"name":"Endocrine journal","volume":" ","pages":"1077-1086"},"PeriodicalIF":2.1000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11778358/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Endocrine journal","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1507/endocrj.EJ24-0147","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/27 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
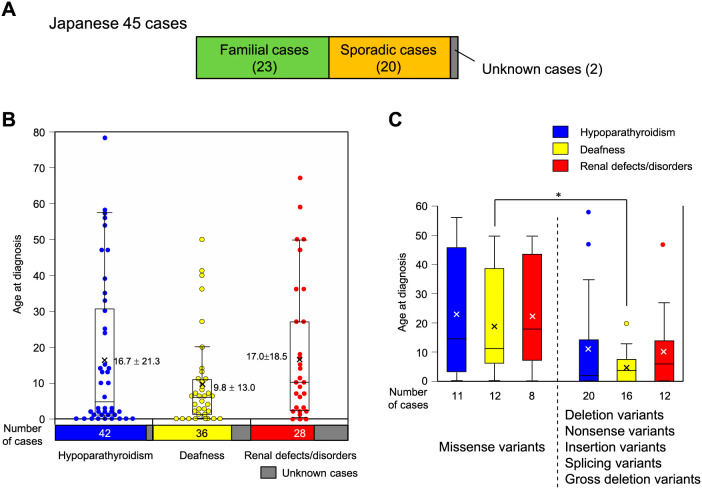
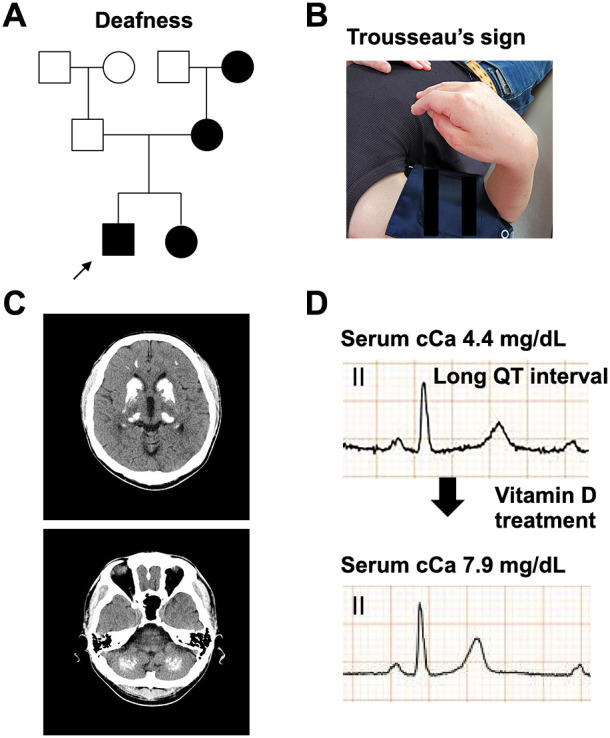
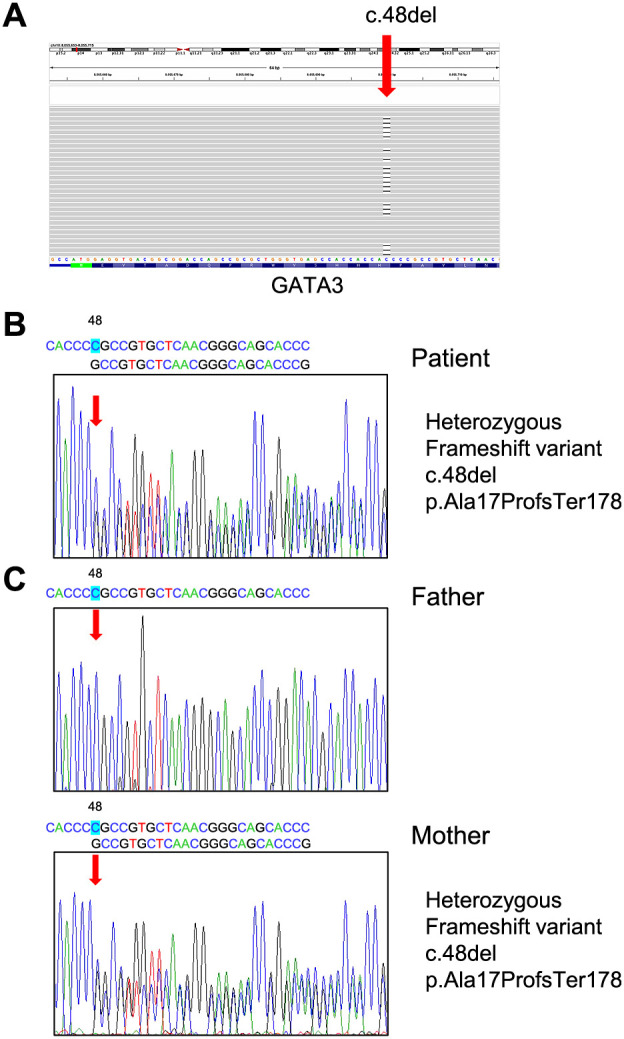
HDR syndrome is an autosomal dominant disorder characterized by hypoparathyroidism (H), deafness (D), and renal dysplasia (R) caused by genetic variants of the GATA3 gene. We present the case of a 38-year-old Japanese man with HDR syndrome who exhibited hypoparathyroidism, sensorineural deafness, renal dysfunction, severe symptomatic hypocalcemia with Chvostek's and Trousseau's signs, and QT prolongation on electrocardiography. He had a family history of deafness and hypocalcemia. Genetic testing revealed a novel GATA3 gene variant at exon 2 (c.48delC), which induces a frameshift resulting in termination at codon 178, causing HDR syndrome. We summarized 45 Japanese cases of HDR syndrome with regard to the mode of onset (familial or sporadic) and the age at diagnosis. In addition, we summarized all previous cases of HDR syndrome with GATA3 gene variants. Mapping of previously reported genetic variants in HDR syndrome revealed that most missense variants were observed at exons 4 and 5 regions in the GATA3 gene. These two regions contain zinc finger domains, demonstrating their functional importance in GATA3 transcription. This review of literature provides a useful reference for diagnosing HDR syndrome and predicting the related future manifestations.
期刊介绍:
Endocrine Journal is an open access, peer-reviewed online journal with a long history. This journal publishes peer-reviewed research articles in multifaceted fields of basic, translational and clinical endocrinology. Endocrine Journal provides a chance to exchange your ideas, concepts and scientific observations in any area of recent endocrinology. Manuscripts may be submitted as Original Articles, Notes, Rapid Communications or Review Articles. We have a rapid reviewing and editorial decision system and pay a special attention to our quick, truly scientific and frequently-citable publication. Please go through the link for author guideline.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
