Machine learning can be as good as maximum likelihood when reconstructing phylogenetic trees and determining the best evolutionary model on four taxon alignments
Nikita Kulikov , Fatemeh Derakhshandeh , Christoph Mayer
{"title":"Machine learning can be as good as maximum likelihood when reconstructing phylogenetic trees and determining the best evolutionary model on four taxon alignments","authors":"Nikita Kulikov , Fatemeh Derakhshandeh , Christoph Mayer","doi":"10.1016/j.ympev.2024.108181","DOIUrl":null,"url":null,"abstract":"<div><p>Phylogenetic tree reconstruction with molecular data is important in many fields of life science research. The gold standard in this discipline is the phylogenetic tree reconstruction based on the Maximum Likelihood method. In this study, we present neural networks to predict the best model of sequence evolution and the correct topology for four sequence alignments of nucleotide or amino acid sequence data. We trained neural networks with different architectures using simulated alignments for a wide range of evolutionary models, model parameters and branch lengths. By comparing the accuracy of model and topology prediction of the trained neural networks with Maximum Likelihood and Neighbour Joining methods, we show that for quartet trees, the neural network classifier outperforms the Neighbour Joining method and is in most cases as good as the Maximum Likelihood method to infer the best model of sequence evolution and the best tree topology. These results are consistent for nucleotide and amino acid sequence data. We also show that our method is superior for model selection than previously published methods based on convolutionary networks. Furthermore, we found that neural network classifiers are much faster than the IQ-TREE implementation of the Maximum Likelihood method. Our results show that neural networks could become a true competitor for the Maximum Likelihood method in phylogenetic reconstructions.</p></div>","PeriodicalId":56109,"journal":{"name":"Molecular Phylogenetics and Evolution","volume":"200 ","pages":"Article 108181"},"PeriodicalIF":3.6000,"publicationDate":"2024-08-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S1055790324001738/pdfft?md5=036b68ef8f10032070e9c004f3188ff9&pid=1-s2.0-S1055790324001738-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Phylogenetics and Evolution","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1055790324001738","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
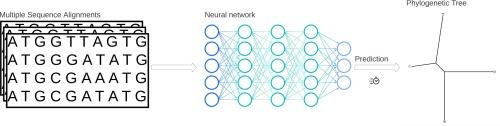
Phylogenetic tree reconstruction with molecular data is important in many fields of life science research. The gold standard in this discipline is the phylogenetic tree reconstruction based on the Maximum Likelihood method. In this study, we present neural networks to predict the best model of sequence evolution and the correct topology for four sequence alignments of nucleotide or amino acid sequence data. We trained neural networks with different architectures using simulated alignments for a wide range of evolutionary models, model parameters and branch lengths. By comparing the accuracy of model and topology prediction of the trained neural networks with Maximum Likelihood and Neighbour Joining methods, we show that for quartet trees, the neural network classifier outperforms the Neighbour Joining method and is in most cases as good as the Maximum Likelihood method to infer the best model of sequence evolution and the best tree topology. These results are consistent for nucleotide and amino acid sequence data. We also show that our method is superior for model selection than previously published methods based on convolutionary networks. Furthermore, we found that neural network classifiers are much faster than the IQ-TREE implementation of the Maximum Likelihood method. Our results show that neural networks could become a true competitor for the Maximum Likelihood method in phylogenetic reconstructions.
期刊介绍:
Molecular Phylogenetics and Evolution is dedicated to bringing Darwin''s dream within grasp - to "have fairly true genealogical trees of each great kingdom of Nature." The journal provides a forum for molecular studies that advance our understanding of phylogeny and evolution, further the development of phylogenetically more accurate taxonomic classifications, and ultimately bring a unified classification for all the ramifying lines of life. Phylogeographic studies will be considered for publication if they offer EXCEPTIONAL theoretical or empirical advances.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
