Qiman Liu , Libin Chen , Manli Zhang , Yujie Hu , Longjiu Cheng
{"title":"Geometrical features, stability and electronic properties of (Cu3Sn)n clusters","authors":"Qiman Liu , Libin Chen , Manli Zhang , Yujie Hu , Longjiu Cheng","doi":"10.1016/j.ica.2024.122340","DOIUrl":null,"url":null,"abstract":"<div><p>Recently, significant advancements have been made in low-entropy Cu<sub>3</sub>Sn catalysts, showcasing their efficient catalytic CO oxidation capabilities. Hence, the atomic models of the Cu<sub>3</sub>Sn are worth established to further investigate their catalytic mechanisms. Here, the structural features and stability of (Cu<sub>3</sub>Sn)<sub>n</sub> clusters (n = 1–6) are investigated using the genetic algorithm combined with the density functional theory (DFT). The results reveal the structural evolution of these clusters from hollow cages to compact icosahedrons, where Cu atoms predominantly tend to grow together, while Sn atoms are dispersed at the edge positions. The <em>E</em><sub>b</sub>, <em>E</em><sub>f</sub> and Δ<sub>2</sub><em>E</em> analyses show that the icosahedral (Cu<sub>3</sub>Sn)<sub>3</sub> has a higher stability than that of its neighbors. The molecular dynamics simulations demonstrates its stability even at 1000 K. The molecular orbitals and density of states reveal that the (Cu<sub>3</sub>Sn)<sub>3</sub> has an 1S<sup>2</sup>1P<sup>6</sup>1D<sup>10</sup>2S<sup>2</sup>1F<sup>1</sup> superatomic electronic configuration. Electrostatic potential surfaces show that (Cu<sub>3</sub>Sn)<sub>n</sub> clusters have significant σ-hole regions at the Cu atomic sites, which can make the C<img>O stretching frequency and bond length have a large red-shift. Moreover, the adsorption energy between the (Cu<sub>3</sub>Sn)<sub>3</sub> and CO is the largest, reaching 1.17 eV. Our work provides inferences to the structural characteristics and adsorptions of the Cu<img>Sn alloys at the atomic level.</p></div>","PeriodicalId":13599,"journal":{"name":"Inorganica Chimica Acta","volume":"573 ","pages":"Article 122340"},"PeriodicalIF":3.2000,"publicationDate":"2024-08-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Inorganica Chimica Acta","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0020169324004316","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
引用次数: 0
Abstract
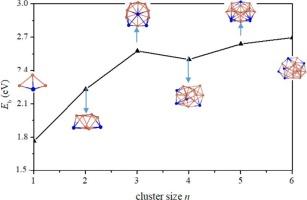
Recently, significant advancements have been made in low-entropy Cu3Sn catalysts, showcasing their efficient catalytic CO oxidation capabilities. Hence, the atomic models of the Cu3Sn are worth established to further investigate their catalytic mechanisms. Here, the structural features and stability of (Cu3Sn)n clusters (n = 1–6) are investigated using the genetic algorithm combined with the density functional theory (DFT). The results reveal the structural evolution of these clusters from hollow cages to compact icosahedrons, where Cu atoms predominantly tend to grow together, while Sn atoms are dispersed at the edge positions. The Eb, Ef and Δ2E analyses show that the icosahedral (Cu3Sn)3 has a higher stability than that of its neighbors. The molecular dynamics simulations demonstrates its stability even at 1000 K. The molecular orbitals and density of states reveal that the (Cu3Sn)3 has an 1S21P61D102S21F1 superatomic electronic configuration. Electrostatic potential surfaces show that (Cu3Sn)n clusters have significant σ-hole regions at the Cu atomic sites, which can make the CO stretching frequency and bond length have a large red-shift. Moreover, the adsorption energy between the (Cu3Sn)3 and CO is the largest, reaching 1.17 eV. Our work provides inferences to the structural characteristics and adsorptions of the CuSn alloys at the atomic level.
期刊介绍:
Inorganica Chimica Acta is an established international forum for all aspects of advanced Inorganic Chemistry. Original papers of high scientific level and interest are published in the form of Articles and Reviews.
Topics covered include:
• chemistry of the main group elements and the d- and f-block metals, including the synthesis, characterization and reactivity of coordination, organometallic, biomimetic, supramolecular coordination compounds, including associated computational studies;
• synthesis, physico-chemical properties, applications of molecule-based nano-scaled clusters and nanomaterials designed using the principles of coordination chemistry, as well as coordination polymers (CPs), metal-organic frameworks (MOFs), metal-organic polyhedra (MPOs);
• reaction mechanisms and physico-chemical investigations computational studies of metalloenzymes and their models;
• applications of inorganic compounds, metallodrugs and molecule-based materials.
Papers composed primarily of structural reports will typically not be considered for publication.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
