Ta Tang, Sulgiye Park, Thomas Peter Devereaux, Yu Lin, Chunjing Jia
{"title":"Molecular geometry specific Monte Carlo simulation of the efficacy of diamond crystal formation from diamondoids","authors":"Ta Tang, Sulgiye Park, Thomas Peter Devereaux, Yu Lin, Chunjing Jia","doi":"10.1038/s42004-024-01261-9","DOIUrl":null,"url":null,"abstract":"Diamondoids are a class of organic molecules with the carbon skeletons isostructural to nano-diamond, and have been shown to be promising precursors for diamond formation. In this work, the formation of diamond crystals from various diamondoid molecule building blocks was studied using our developed molecular geometry specific Monte Carlo method. We maintained the internal carbon skeletons of the diamondoid molecules, and investigated how the carbon-carbon bonds form between diamondoid molecules and how efficient the process is to form diamond crystals. The simulations show that higher diamondoid molecules can produce structures closer to a diamond crystal compared with lower diamondoid molecules. Specifically, using higher diamondoid molecules, larger bulk diamond crystals are formed with fewer vacancies. The higher propensity of certain diamondoids to form diamond crystals reveals insights into the microscopic processes of diamond formation under high-pressure high-temperature conditions. Diamondoids are a series of hydrogen-terminated nanometer-sized hydrocarbons that can be used to synthesize high-quality diamond crystals. Here, the authors use Monte Carlo simulations to study the potentials of different diamondoids in constructing diamond crystals with the assumption that the carbon skeletons keep intact, and find that higher diamondoid molecules are most suitable.","PeriodicalId":10529,"journal":{"name":"Communications Chemistry","volume":" ","pages":"1-5"},"PeriodicalIF":6.2000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s42004-024-01261-9.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Communications Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.nature.com/articles/s42004-024-01261-9","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
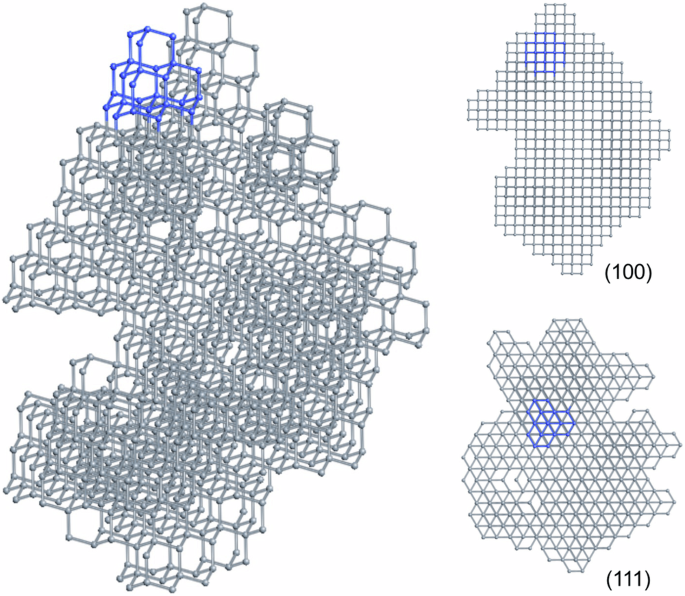
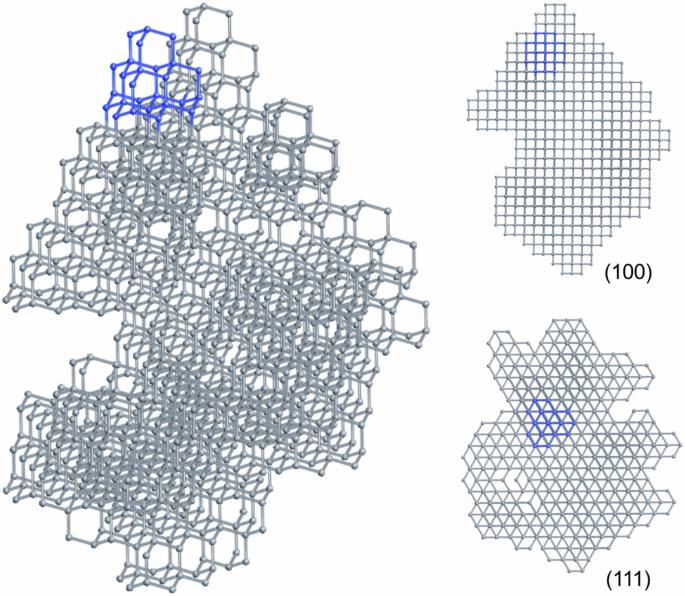
Diamondoids are a class of organic molecules with the carbon skeletons isostructural to nano-diamond, and have been shown to be promising precursors for diamond formation. In this work, the formation of diamond crystals from various diamondoid molecule building blocks was studied using our developed molecular geometry specific Monte Carlo method. We maintained the internal carbon skeletons of the diamondoid molecules, and investigated how the carbon-carbon bonds form between diamondoid molecules and how efficient the process is to form diamond crystals. The simulations show that higher diamondoid molecules can produce structures closer to a diamond crystal compared with lower diamondoid molecules. Specifically, using higher diamondoid molecules, larger bulk diamond crystals are formed with fewer vacancies. The higher propensity of certain diamondoids to form diamond crystals reveals insights into the microscopic processes of diamond formation under high-pressure high-temperature conditions. Diamondoids are a series of hydrogen-terminated nanometer-sized hydrocarbons that can be used to synthesize high-quality diamond crystals. Here, the authors use Monte Carlo simulations to study the potentials of different diamondoids in constructing diamond crystals with the assumption that the carbon skeletons keep intact, and find that higher diamondoid molecules are most suitable.
期刊介绍:
Communications Chemistry is an open access journal from Nature Research publishing high-quality research, reviews and commentary in all areas of the chemical sciences. Research papers published by the journal represent significant advances bringing new chemical insight to a specialized area of research. We also aim to provide a community forum for issues of importance to all chemists, regardless of sub-discipline.


 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
