Eric Jonathan Maciel-Cruz, Luis Eduardo Figuera-Villanueva, Liliana Gómez-Flores-Ramos, Rubiceli Hernández-Peña, Martha Patricia Gallegos-Arreola
{"title":"<i>In-Silico</i> Method for Predicting Pathogenic Missense Variants Using Online Tools: AURKA Gene as a Model.","authors":"Eric Jonathan Maciel-Cruz, Luis Eduardo Figuera-Villanueva, Liliana Gómez-Flores-Ramos, Rubiceli Hernández-Peña, Martha Patricia Gallegos-Arreola","doi":"10.30498/ijb.2024.413800.3787","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong><i>In-silico</i> analysis provides a fast, simple, and cost-free method for identifying potentially pathogenic single nucleotide variants.</p><p><strong>Objective: </strong>To propose a simple and relatively fast method for the prediction of variant pathogenicity using free online <i>in-silico</i> (IS) tools with <i>AURKA</i> gene as a model.</p><p><strong>Materials and methods: </strong>We aim to propose a methodology to predict variants with high pathogenic potential using computational analysis, using <i>AURKA</i> gene as model. We predicted a protein model and analyzed 209 out of 64,369 <i>AURKA</i> variants obtained from Ensembl database. We used bioinformatic tools to predict pathogenicity. The results were compared through the VarSome website, which includes its own pathogenicity score and the American College of Medical Genetics (ACMG) classification.</p><p><strong>Results: </strong>Out of the 209 analyzed variants, 16 were considered pathogenic, and 13 were located in the catalytic domain. The most frequent protein changes were size and hydrophobicity modifications of amino acids. Proline and Glycine amino acid substitutions were the most frequent changes predicted as pathogenic. These bioinformatic tools predicted functional changes, such as protein up or down-regulation, gain or loss of molecule interactions, and structural protein modifications. When compared to the ACMG classification, 10 out of 16 variants were considered likely pathogenic, with 7 out of 10 changes at Proline/Glycine substitutions.</p><p><strong>Conclusion: </strong>This method allows quick and cost-free bulk variant screening to identify variants with pathogenic potential for further association and/or functional studies.</p>","PeriodicalId":14492,"journal":{"name":"Iranian Journal of Biotechnology","volume":"22 2","pages":"e3787"},"PeriodicalIF":1.5000,"publicationDate":"2024-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11364922/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Iranian Journal of Biotechnology","FirstCategoryId":"5","ListUrlMain":"https://doi.org/10.30498/ijb.2024.413800.3787","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: In-silico analysis provides a fast, simple, and cost-free method for identifying potentially pathogenic single nucleotide variants.
Objective: To propose a simple and relatively fast method for the prediction of variant pathogenicity using free online in-silico (IS) tools with AURKA gene as a model.
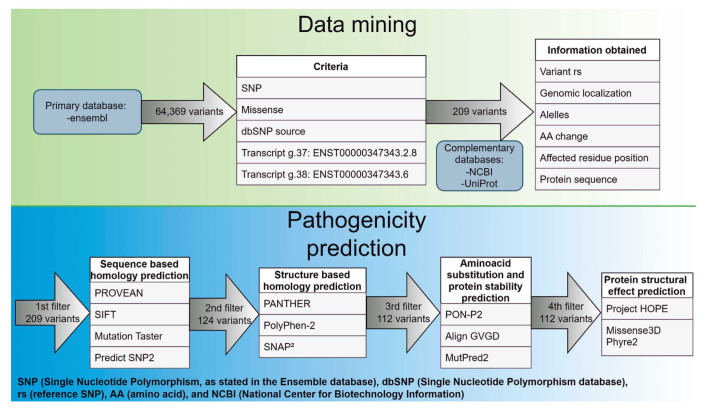
Materials and methods: We aim to propose a methodology to predict variants with high pathogenic potential using computational analysis, using AURKA gene as model. We predicted a protein model and analyzed 209 out of 64,369 AURKA variants obtained from Ensembl database. We used bioinformatic tools to predict pathogenicity. The results were compared through the VarSome website, which includes its own pathogenicity score and the American College of Medical Genetics (ACMG) classification.
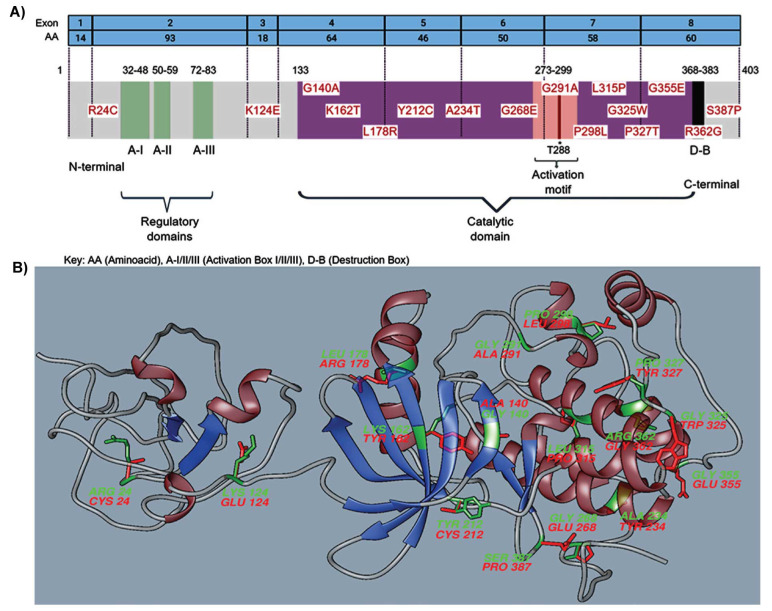
Results: Out of the 209 analyzed variants, 16 were considered pathogenic, and 13 were located in the catalytic domain. The most frequent protein changes were size and hydrophobicity modifications of amino acids. Proline and Glycine amino acid substitutions were the most frequent changes predicted as pathogenic. These bioinformatic tools predicted functional changes, such as protein up or down-regulation, gain or loss of molecule interactions, and structural protein modifications. When compared to the ACMG classification, 10 out of 16 variants were considered likely pathogenic, with 7 out of 10 changes at Proline/Glycine substitutions.
Conclusion: This method allows quick and cost-free bulk variant screening to identify variants with pathogenic potential for further association and/or functional studies.
期刊介绍:
Iranian Journal of Biotechnology (IJB) is published quarterly by the National Institute of Genetic Engineering and Biotechnology. IJB publishes original scientific research papers in the broad area of Biotechnology such as, Agriculture, Animal and Marine Sciences, Basic Sciences, Bioinformatics, Biosafety and Bioethics, Environment, Industry and Mining and Medical Sciences.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
