{"title":"Context-adjusted proportion of singletons (CAPS): a novel metric for assessing negative selection in the human genome.","authors":"Mikhail Gudkov, Loïc Thibaut, Eleni Giannoulatou","doi":"10.1093/nargab/lqae111","DOIUrl":null,"url":null,"abstract":"<p><p>Interpretation of genetic variants remains challenging, partly due to the lack of well-established ways of determining the potential pathogenicity of genetic variation, especially for understudied classes of variants. Addressing this, population genetics methods offer a practical solution by evaluating variant effects through human population distributions. Negative selection influences the ratio of singleton variants and can serve as a proxy for deleteriousness, as exemplified by the Mutability-Adjusted Proportion of Singletons (MAPS) metric. However, MAPS is sensitive to the calibration of the singletons-by-mutability linear model, which results in biased estimates for certain variant classes. Building up on the methodology used in MAPS, we introduce the Context-Adjusted Proportion of Singletons (CAPS) metric for assessing negative selection in the human genome. CAPS produces corrected estimates with more accurate confidence intervals by eliminating the mutability layer in the model. Retaining the advantageous features of MAPS, CAPS emerges as a robust and reliable tool. We believe that CAPS has the potential to enhance the identification of new disease-variant associations in clinical and research settings, offering improved accuracy in assessing negative selection for diverse SNV classes.</p>","PeriodicalId":33994,"journal":{"name":"NAR Genomics and Bioinformatics","volume":"6 3","pages":"lqae111"},"PeriodicalIF":2.8000,"publicationDate":"2024-08-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11358819/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NAR Genomics and Bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/nargab/lqae111","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
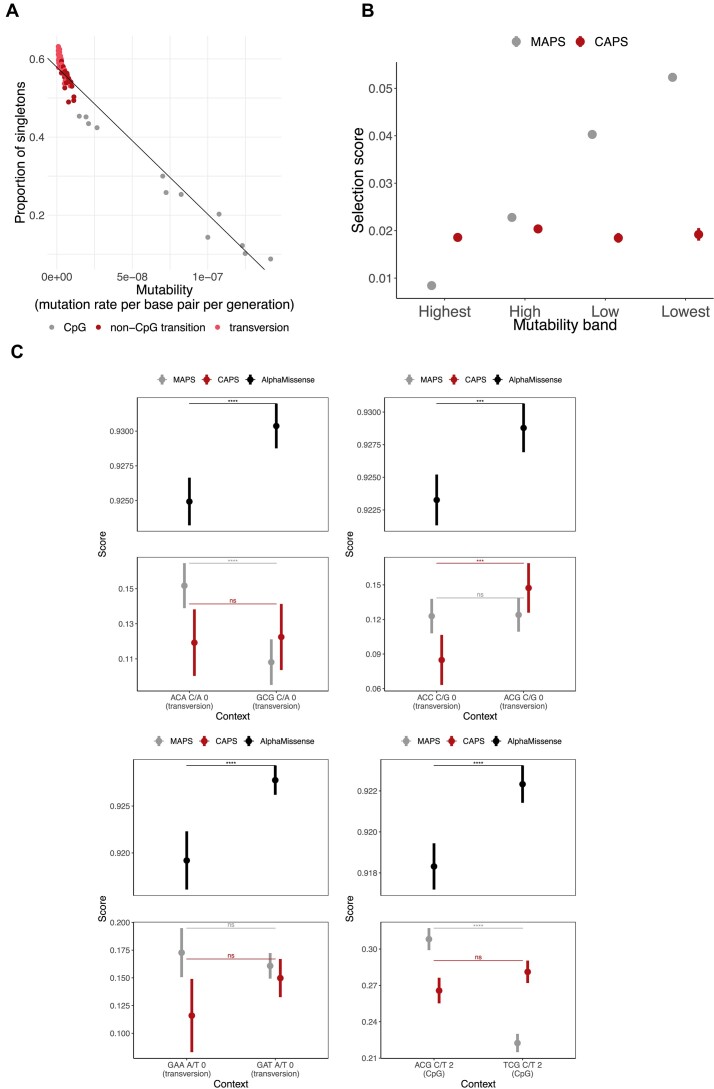
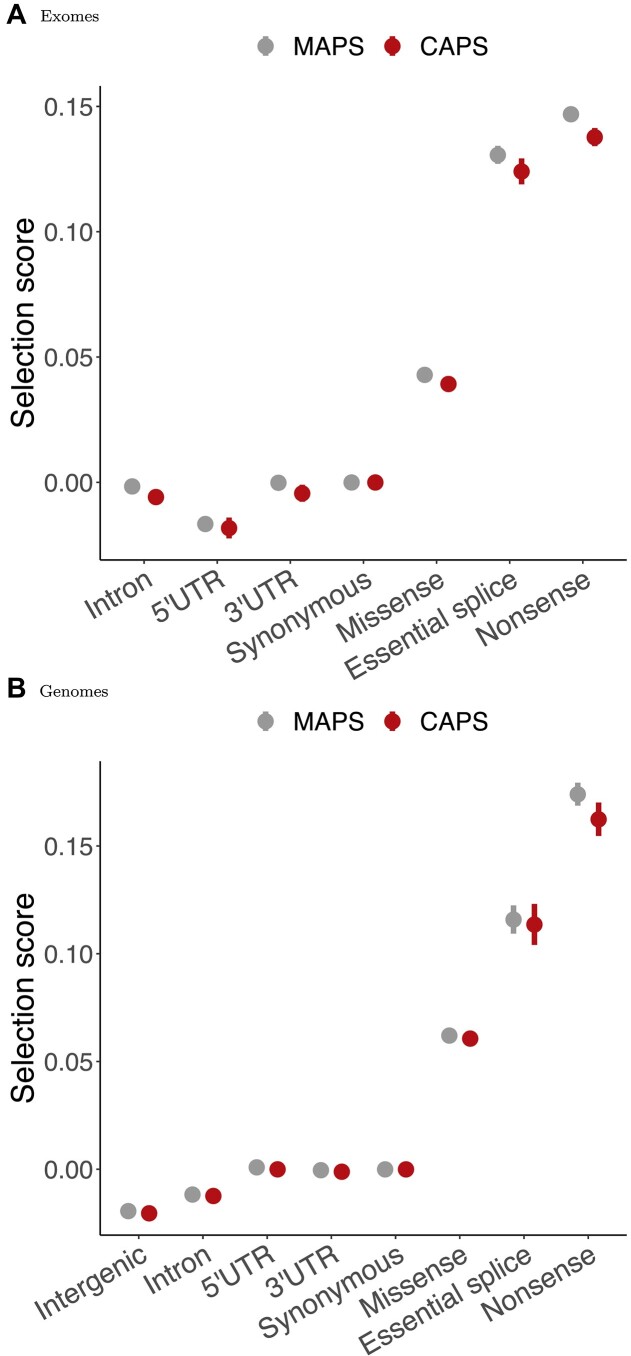
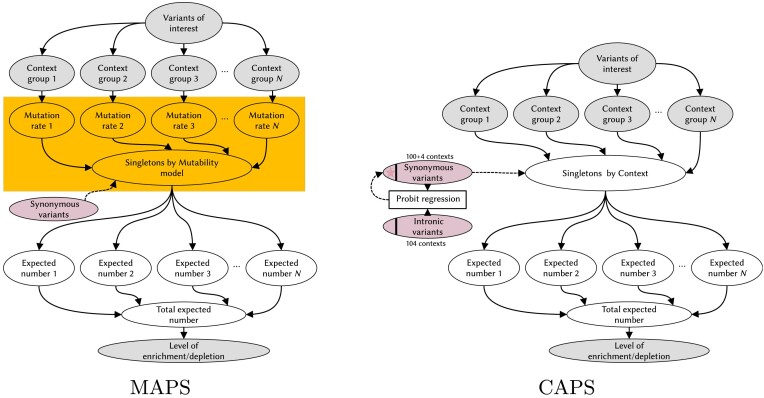
Interpretation of genetic variants remains challenging, partly due to the lack of well-established ways of determining the potential pathogenicity of genetic variation, especially for understudied classes of variants. Addressing this, population genetics methods offer a practical solution by evaluating variant effects through human population distributions. Negative selection influences the ratio of singleton variants and can serve as a proxy for deleteriousness, as exemplified by the Mutability-Adjusted Proportion of Singletons (MAPS) metric. However, MAPS is sensitive to the calibration of the singletons-by-mutability linear model, which results in biased estimates for certain variant classes. Building up on the methodology used in MAPS, we introduce the Context-Adjusted Proportion of Singletons (CAPS) metric for assessing negative selection in the human genome. CAPS produces corrected estimates with more accurate confidence intervals by eliminating the mutability layer in the model. Retaining the advantageous features of MAPS, CAPS emerges as a robust and reliable tool. We believe that CAPS has the potential to enhance the identification of new disease-variant associations in clinical and research settings, offering improved accuracy in assessing negative selection for diverse SNV classes.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
