Agustin Hidalgo-Gutierrez PhD, Jonathan Shintaku, Javier Ramon, Eliana Barriocanal-Casado, Alba Pesini, Russell P. Saneto, Gloria Garrabou, Jose Cesar Milisenda, Ana Matas-Garcia, Laura Gort, Olatz Ugarteburu, Yue Gu, Lahari Koganti, Tian Wang, Saba Tadesse, Megi Meneri, Monica Sciacco, Shuang Wang, Kurenai Tanji, Marshall S. Horwitz, Michael O. Dorschner, Mahesh Mansukhani, Giacomo Pietro Comi, Dario Ronchi, Ramon Marti, Antonia Ribes, Frederic Tort, Michio Hirano MD
{"title":"Guanylate Kinase 1 Deficiency: A Novel and Potentially Treatable Mitochondrial DNA Depletion/Deletions Disease","authors":"Agustin Hidalgo-Gutierrez PhD, Jonathan Shintaku, Javier Ramon, Eliana Barriocanal-Casado, Alba Pesini, Russell P. Saneto, Gloria Garrabou, Jose Cesar Milisenda, Ana Matas-Garcia, Laura Gort, Olatz Ugarteburu, Yue Gu, Lahari Koganti, Tian Wang, Saba Tadesse, Megi Meneri, Monica Sciacco, Shuang Wang, Kurenai Tanji, Marshall S. Horwitz, Michael O. Dorschner, Mahesh Mansukhani, Giacomo Pietro Comi, Dario Ronchi, Ramon Marti, Antonia Ribes, Frederic Tort, Michio Hirano MD","doi":"10.1002/ana.27071","DOIUrl":null,"url":null,"abstract":"<div>\n \n <section>\n \n <h3> Objective</h3>\n \n <p>Mitochondrial DNA (mtDNA) depletion/deletions syndrome (MDDS) comprises a group of diseases caused by primary autosomal defects of mtDNA maintenance. Our objective was to study the etiology of MDDS in 4 patients who lack pathogenic variants in known genetic causes.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>Whole exome sequencing of the probands was performed to identify pathogenic variants. We validated the mitochondrial defect by analyzing mtDNA, mitochondrial dNTP pools, respiratory chain activities, and GUK1 activity. To confirm pathogenicity of GUK1 deficiency, we expressed 2 GUK1 isoforms in patient cells.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>We identified biallelic <i>GUK1</i> pathogenic variants in all 4 probands who presented with ptosis, ophthalmoparesis, and myopathic proximal limb weakness, as well as variable hepatopathy and altered T-lymphocyte profiles. Muscle biopsies from all probands showed mtDNA depletion, deletions, or both, as well as reduced activities of mitochondrial respiratory chain enzymes. <i>GUK1</i> encodes guanylate kinase, originally identified as a cytosolic enzyme. Long and short isoforms of GUK1 exist. We observed that the long isoform is intramitochondrial and the short is cytosolic. In probands’ fibroblasts, we noted decreased GUK1 activity causing unbalanced mitochondrial dNTP pools and mtDNA depletion in both replicating and quiescent fibroblasts indicating that GUK1 deficiency impairs de novo and salvage nucleotide pathways. Proband fibroblasts treated with deoxyguanosine and/or forodesine, a purine phosphatase inhibitor, ameliorated mtDNA depletion, indicating potential pharmacological therapies.</p>\n </section>\n \n <section>\n \n <h3> Interpretation</h3>\n \n <p>Primary GUK1 deficiency is a new and potentially treatable cause of MDDS. The cytosolic isoform of GUK1 may contribute to the T-lymphocyte abnormality, which has not been observed in other MDDS disorders. ANN NEUROL 2024;96:1209–1224</p>\n </section>\n </div>","PeriodicalId":127,"journal":{"name":"Annals of Neurology","volume":"96 6","pages":"1209-1224"},"PeriodicalIF":7.7000,"publicationDate":"2024-09-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11563867/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Annals of Neurology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ana.27071","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Objective
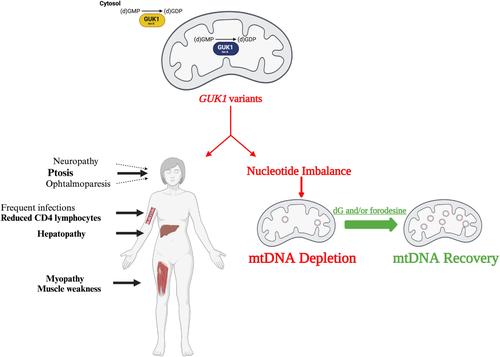
Mitochondrial DNA (mtDNA) depletion/deletions syndrome (MDDS) comprises a group of diseases caused by primary autosomal defects of mtDNA maintenance. Our objective was to study the etiology of MDDS in 4 patients who lack pathogenic variants in known genetic causes.
Methods
Whole exome sequencing of the probands was performed to identify pathogenic variants. We validated the mitochondrial defect by analyzing mtDNA, mitochondrial dNTP pools, respiratory chain activities, and GUK1 activity. To confirm pathogenicity of GUK1 deficiency, we expressed 2 GUK1 isoforms in patient cells.
Results
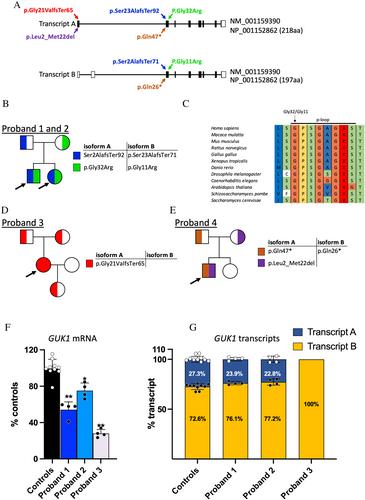
We identified biallelic GUK1 pathogenic variants in all 4 probands who presented with ptosis, ophthalmoparesis, and myopathic proximal limb weakness, as well as variable hepatopathy and altered T-lymphocyte profiles. Muscle biopsies from all probands showed mtDNA depletion, deletions, or both, as well as reduced activities of mitochondrial respiratory chain enzymes. GUK1 encodes guanylate kinase, originally identified as a cytosolic enzyme. Long and short isoforms of GUK1 exist. We observed that the long isoform is intramitochondrial and the short is cytosolic. In probands’ fibroblasts, we noted decreased GUK1 activity causing unbalanced mitochondrial dNTP pools and mtDNA depletion in both replicating and quiescent fibroblasts indicating that GUK1 deficiency impairs de novo and salvage nucleotide pathways. Proband fibroblasts treated with deoxyguanosine and/or forodesine, a purine phosphatase inhibitor, ameliorated mtDNA depletion, indicating potential pharmacological therapies.
Interpretation
Primary GUK1 deficiency is a new and potentially treatable cause of MDDS. The cytosolic isoform of GUK1 may contribute to the T-lymphocyte abnormality, which has not been observed in other MDDS disorders. ANN NEUROL 2024;96:1209–1224
期刊介绍:
Annals of Neurology publishes original articles with potential for high impact in understanding the pathogenesis, clinical and laboratory features, diagnosis, treatment, outcomes and science underlying diseases of the human nervous system. Articles should ideally be of broad interest to the academic neurological community rather than solely to subspecialists in a particular field. Studies involving experimental model system, including those in cell and organ cultures and animals, of direct translational relevance to the understanding of neurological disease are also encouraged.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
