Phenotypic spectrum of iron-sulfur cluster assembly gene IBA57 mutations: c.286 T > C identified as a hotspot mutation in Chinese patients with a stable natural history
{"title":"Phenotypic spectrum of iron-sulfur cluster assembly gene IBA57 mutations: c.286 T > C identified as a hotspot mutation in Chinese patients with a stable natural history","authors":"Huafang Jiang, Chaolong Xu, Ruoyu Duan, Zhimei Liu, Xiaotun Ren, Jiuwei Li, Chunhong Chen, Hongmei Wang, Tongli Han, Xiaojuan Tian, Xin Duan, Minhan Song, Tongyue Li, Fang Fang","doi":"10.1038/s10038-024-01291-0","DOIUrl":null,"url":null,"abstract":"Mutations in IBA57 disrupt iron-sulfur clusters maturation, causing a rare mitochondrial disease. Clinical manifestations vary from neonatal lethality to childhood-onset spastic paraparesis, yet the ethnic heterogeneity and natural history remain unclear, necessitating further exploration. This study aimed to delineate the genotype-phenotype correlation of IBA57 mutations by analyzing diverse clinical presentations. We report 11 Chinese patients and include literature-reported cases, totaling 61 patients enrolled for analysis. Clinical, neuroimaging, genetic, and disease progression information were collected. Among these, 46 presented as multiple mitochondrial dysfunctions syndrome 3 (MMDS3), with 58.7% originating from Chinese population. Based on disease course, we propose three clinical subtypes: neonatal, infant and childhood subtypes. Neonatal cases universally displayed hypotonia and respiratory distress at presentation, deceased within three months. Most infancy and childhood cases exhibited developmental regression and impaired motor function. Cavitating leukoencephalopathy was a typical neuroimaging finding in MMDS3 patients. The c.286 T > C mutation was reported in 85.2% of Chinese patients. A significantly lower mortality rate was observed compared to the non-Chinese group (P = 0.002), with a survival rate exceeding 90% at 5 years, indicating a relatively stable disease progression. Fifteen cases from three families manifested the spastic paraplegia 74 phenotype, demonstrating normal development before onset, with common clinical manifestations including spastic paraplegia (14/15), visual impairment (10/13), and peripheral neuropathy (9/13). In conclusion, this study indicates a hotspot mutation in Chinese and analyses the disease progression with different clinical subtypes.","PeriodicalId":16077,"journal":{"name":"Journal of Human Genetics","volume":"70 1","pages":"25-32"},"PeriodicalIF":2.5000,"publicationDate":"2024-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s10038-024-01291-0","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
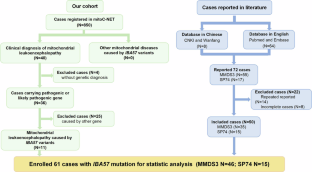
Mutations in IBA57 disrupt iron-sulfur clusters maturation, causing a rare mitochondrial disease. Clinical manifestations vary from neonatal lethality to childhood-onset spastic paraparesis, yet the ethnic heterogeneity and natural history remain unclear, necessitating further exploration. This study aimed to delineate the genotype-phenotype correlation of IBA57 mutations by analyzing diverse clinical presentations. We report 11 Chinese patients and include literature-reported cases, totaling 61 patients enrolled for analysis. Clinical, neuroimaging, genetic, and disease progression information were collected. Among these, 46 presented as multiple mitochondrial dysfunctions syndrome 3 (MMDS3), with 58.7% originating from Chinese population. Based on disease course, we propose three clinical subtypes: neonatal, infant and childhood subtypes. Neonatal cases universally displayed hypotonia and respiratory distress at presentation, deceased within three months. Most infancy and childhood cases exhibited developmental regression and impaired motor function. Cavitating leukoencephalopathy was a typical neuroimaging finding in MMDS3 patients. The c.286 T > C mutation was reported in 85.2% of Chinese patients. A significantly lower mortality rate was observed compared to the non-Chinese group (P = 0.002), with a survival rate exceeding 90% at 5 years, indicating a relatively stable disease progression. Fifteen cases from three families manifested the spastic paraplegia 74 phenotype, demonstrating normal development before onset, with common clinical manifestations including spastic paraplegia (14/15), visual impairment (10/13), and peripheral neuropathy (9/13). In conclusion, this study indicates a hotspot mutation in Chinese and analyses the disease progression with different clinical subtypes.
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
