Joshua S Weinstock, Sharjeel A Chaudhry, Maria Ioannou, Maria Viskadourou, Paula Reventun, Yasminka A Jakubek, L Alexander Liggett, Cecelia Laurie, Jai G Broome, Alyna Khan, Kent D Taylor, Xiuqing Guo, Patricia A Peyser, Eric Boerwinkle, Nathalie Chami, Eimear E Kenny, Ruth J Loos, Bruce M Psaty, Tracy P Russell, Jennifer A Brody, Jeong H Yun, Michael H Cho, Ramachandran S Vasan, Sharon L Kardia, Jennifer A Smith, Laura M Raffield, Aurelian Bidulescu, Emily O'Brien, Mariza de Andrade, Jerome I Rotter, Stephen S Rich, Russell P Tracy, Yii Der Ida Chen, C Charles Gu, Chao A Hsiung, Charles Kooperberg, Bernhard Haring, Rami Nassir, Rasika Mathias, Alex Reiner, Vijay Sankaran, Charles J Lowenstein, Thomas W Blackwell, Goncalo R Abecasis, Albert V Smith, Hyun M Kang, Pradeep Natarajan, Siddhartha Jaiswal, Alexander Bick, Wendy S Post, Paul Scheet, Paul Auer, Theodoros Karantanos, Alexis Battle, Marios Arvanitis
{"title":"The Genetic Determinants and Genomic Consequences of Non-Leukemogenic Somatic Point Mutations.","authors":"Joshua S Weinstock, Sharjeel A Chaudhry, Maria Ioannou, Maria Viskadourou, Paula Reventun, Yasminka A Jakubek, L Alexander Liggett, Cecelia Laurie, Jai G Broome, Alyna Khan, Kent D Taylor, Xiuqing Guo, Patricia A Peyser, Eric Boerwinkle, Nathalie Chami, Eimear E Kenny, Ruth J Loos, Bruce M Psaty, Tracy P Russell, Jennifer A Brody, Jeong H Yun, Michael H Cho, Ramachandran S Vasan, Sharon L Kardia, Jennifer A Smith, Laura M Raffield, Aurelian Bidulescu, Emily O'Brien, Mariza de Andrade, Jerome I Rotter, Stephen S Rich, Russell P Tracy, Yii Der Ida Chen, C Charles Gu, Chao A Hsiung, Charles Kooperberg, Bernhard Haring, Rami Nassir, Rasika Mathias, Alex Reiner, Vijay Sankaran, Charles J Lowenstein, Thomas W Blackwell, Goncalo R Abecasis, Albert V Smith, Hyun M Kang, Pradeep Natarajan, Siddhartha Jaiswal, Alexander Bick, Wendy S Post, Paul Scheet, Paul Auer, Theodoros Karantanos, Alexis Battle, Marios Arvanitis","doi":"10.1101/2024.08.22.24312319","DOIUrl":null,"url":null,"abstract":"<p><p>Clonal hematopoiesis (CH) is defined by the expansion of a lineage of genetically identical cells in blood. Genetic lesions that confer a fitness advantage, such as point mutations or mosaic chromosomal alterations (mCAs) in genes associated with hematologic malignancy, are frequent mediators of CH. However, recent analyses of both single cell-derived colonies of hematopoietic cells and population sequencing cohorts have revealed CH frequently occurs in the absence of known driver genetic lesions. To characterize CH without known driver genetic lesions, we used 51,399 deeply sequenced whole genomes from the NHLBI TOPMed sequencing initiative to perform simultaneous germline and somatic mutation analyses among individuals without leukemogenic point mutations (LPM), which we term CH-LPMneg. We quantified CH by estimating the total mutation burden. Because estimating somatic mutation burden without a paired-tissue sample is challenging, we developed a novel statistical method, the Genomic and Epigenomic informed Mutation (GEM) rate, that uses external genomic and epigenomic data sources to distinguish artifactual signals from true somatic mutations. We performed a genome-wide association study of GEM to discover the germline determinants of CH-LPMneg. After fine-mapping and variant-to-gene analyses, we identified seven genes associated with CH-LPMneg (<i>TCL1A, TERT, SMC4, NRIP1, PRDM16</i>, <i>MSRA</i>, <i>SCARB1</i>), and one locus associated with a sex-associated mutation pathway (<i>SRGAP2C)</i>. We performed a secondary analysis excluding individuals with mCAs, finding that the genetic architecture was largely unaffected by their inclusion. Functional analyses of <i>SMC4</i> and <i>NRIP1</i> implicated altered HSC self-renewal and proliferation as the primary mediator of mutation burden in blood. We then performed comprehensive multi-tissue transcriptomic analyses, finding that the expression levels of 404 genes are associated with GEM. Finally, we performed phenotypic association meta-analyses across four cohorts, finding that GEM is associated with increased white blood cell count and increased risk for incident peripheral artery disease, but is not significantly associated with incident stroke or coronary disease events. Overall, we develop GEM for quantifying mutation burden from WGS without a paired-tissue sample and use GEM to discover the genetic, genomic, and phenotypic correlates of CH-LPMneg.</p>","PeriodicalId":94281,"journal":{"name":"medRxiv : the preprint server for health sciences","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-08-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11370504/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"medRxiv : the preprint server for health sciences","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/2024.08.22.24312319","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
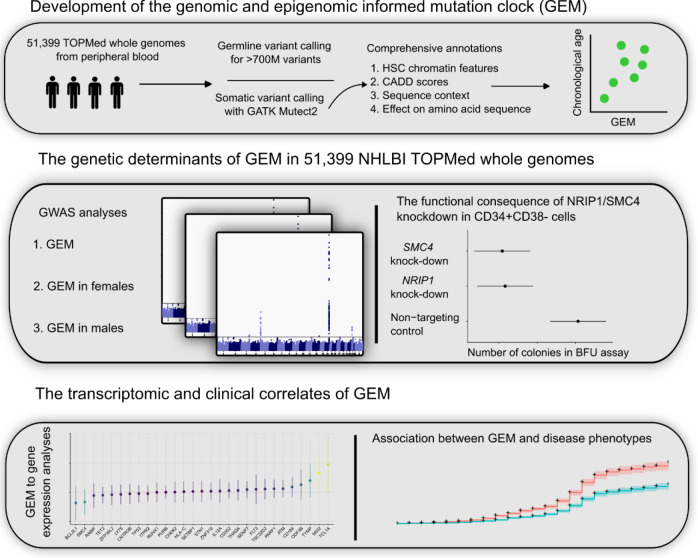
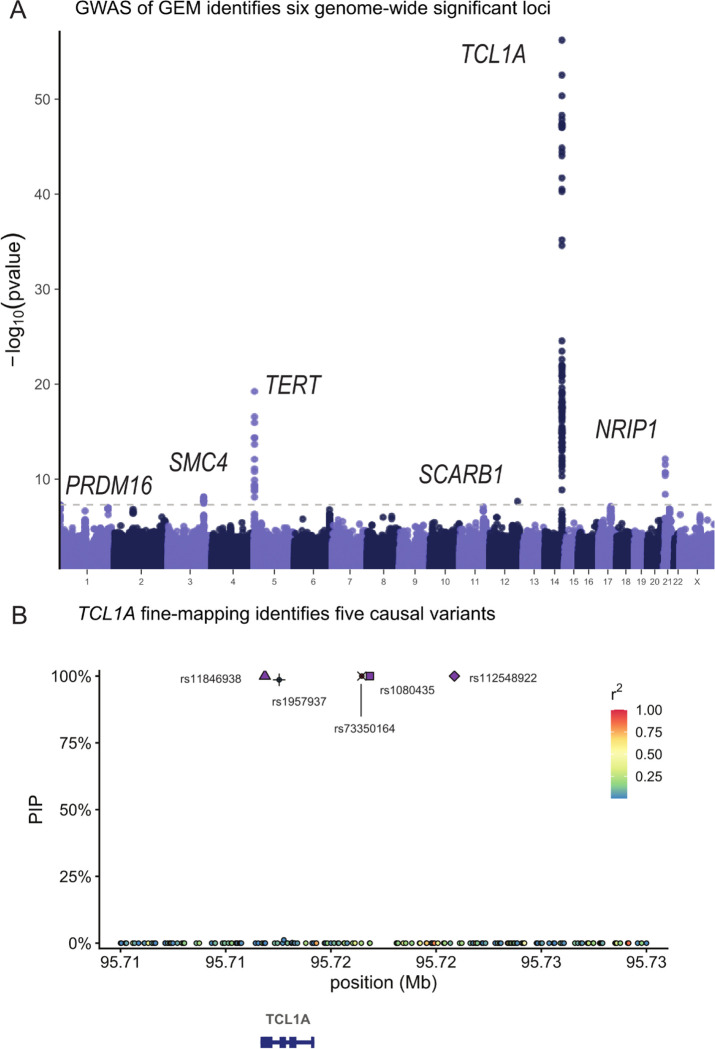
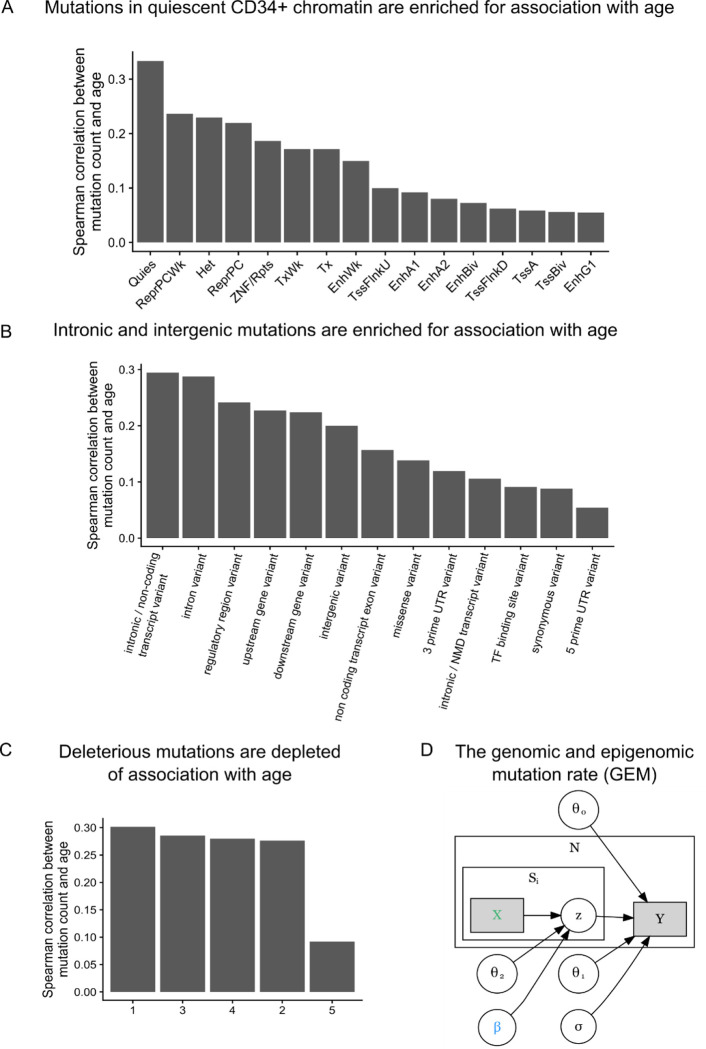
Clonal hematopoiesis (CH) is defined by the expansion of a lineage of genetically identical cells in blood. Genetic lesions that confer a fitness advantage, such as point mutations or mosaic chromosomal alterations (mCAs) in genes associated with hematologic malignancy, are frequent mediators of CH. However, recent analyses of both single cell-derived colonies of hematopoietic cells and population sequencing cohorts have revealed CH frequently occurs in the absence of known driver genetic lesions. To characterize CH without known driver genetic lesions, we used 51,399 deeply sequenced whole genomes from the NHLBI TOPMed sequencing initiative to perform simultaneous germline and somatic mutation analyses among individuals without leukemogenic point mutations (LPM), which we term CH-LPMneg. We quantified CH by estimating the total mutation burden. Because estimating somatic mutation burden without a paired-tissue sample is challenging, we developed a novel statistical method, the Genomic and Epigenomic informed Mutation (GEM) rate, that uses external genomic and epigenomic data sources to distinguish artifactual signals from true somatic mutations. We performed a genome-wide association study of GEM to discover the germline determinants of CH-LPMneg. After fine-mapping and variant-to-gene analyses, we identified seven genes associated with CH-LPMneg (TCL1A, TERT, SMC4, NRIP1, PRDM16, MSRA, SCARB1), and one locus associated with a sex-associated mutation pathway (SRGAP2C). We performed a secondary analysis excluding individuals with mCAs, finding that the genetic architecture was largely unaffected by their inclusion. Functional analyses of SMC4 and NRIP1 implicated altered HSC self-renewal and proliferation as the primary mediator of mutation burden in blood. We then performed comprehensive multi-tissue transcriptomic analyses, finding that the expression levels of 404 genes are associated with GEM. Finally, we performed phenotypic association meta-analyses across four cohorts, finding that GEM is associated with increased white blood cell count and increased risk for incident peripheral artery disease, but is not significantly associated with incident stroke or coronary disease events. Overall, we develop GEM for quantifying mutation burden from WGS without a paired-tissue sample and use GEM to discover the genetic, genomic, and phenotypic correlates of CH-LPMneg.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
