Signe Mosegaard, Krishna S. Twayana, Simone W. Denis, Jeffrey Kroon, Bauke V. Schomakers, Michel van Weeghel, Riekelt H. Houtkooper, Rikke K. J. Olsen, Christian K. Holm
{"title":"Human inborn errors of long-chain fatty acid oxidation show impaired inflammatory responses to TLR4-ligand LPS","authors":"Signe Mosegaard, Krishna S. Twayana, Simone W. Denis, Jeffrey Kroon, Bauke V. Schomakers, Michel van Weeghel, Riekelt H. Houtkooper, Rikke K. J. Olsen, Christian K. Holm","doi":"10.1096/fba.2024-00060","DOIUrl":null,"url":null,"abstract":"<p>Stimulation of mammalian cells with inflammatory inducers such as lipopolysaccharide (LPS) leads to alterations in activity of central cellular metabolic pathways. Interestingly, these metabolic changes seem to be important for subsequent release of pro-inflammatory cytokines. This has become particularly clear for enzymes of tricarboxylic acid (TCA) cycle such as succinate dehydrogenase (<i>SDH</i>). LPS leads to inhibition of SDH activity and accumulation of succinate to enhance the LPS-induced formation of IL-1β. If enzymes involved in beta-oxidation of fatty acids are important for sufficient responses to LPS is currently not clear. Using cells from various patients with inborn long-chain fatty acid oxidation disorders (lcFAOD), we report that disease-causing deleterious variants of Electron Transfer Flavoprotein Dehydrogenase (<i>ETFDH</i>) and of Very Long Chain Acyl-CoA Dehydrogenase (<i>ACADVL</i>), both cause insufficient inflammatory responses to stimulation with LPS. The insufficiencies included reduced TLR4 expression levels, impaired TLR4 signaling, and reduced or absent induction of pro-inflammatory cytokines such as IL-6. The insufficient responses to LPS were reproduced in cells from healthy controls by targeted loss-of-function of either <i>ETFDH</i> or <i>ACADVL,</i> supporting that the deleterious <i>ETFDH</i> and <i>ACADVL</i> variants cause the attenuated responses to LPS. <i>ETFDH</i> and <i>ACADVL</i> encode two distinct enzymes both involved in fatty acid beta-oxidation, and patients with these deficiencies cannot sufficiently metabolize long-chain fatty acids. We report that genes important for beta-oxidation of long-chain fatty acids are also important for inflammatory responses to an acute immunogen trigger like LPS, which may have important implications for understanding infection and other metabolic stress induced disease pathology in lcFAODs.</p>","PeriodicalId":12093,"journal":{"name":"FASEB bioAdvances","volume":"6 9","pages":"337-350"},"PeriodicalIF":2.4000,"publicationDate":"2024-08-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1096/fba.2024-00060","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"FASEB bioAdvances","FirstCategoryId":"1085","ListUrlMain":"https://faseb.onlinelibrary.wiley.com/doi/10.1096/fba.2024-00060","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
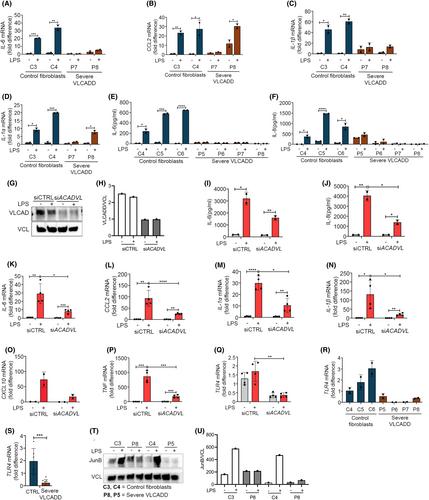
Stimulation of mammalian cells with inflammatory inducers such as lipopolysaccharide (LPS) leads to alterations in activity of central cellular metabolic pathways. Interestingly, these metabolic changes seem to be important for subsequent release of pro-inflammatory cytokines. This has become particularly clear for enzymes of tricarboxylic acid (TCA) cycle such as succinate dehydrogenase (SDH). LPS leads to inhibition of SDH activity and accumulation of succinate to enhance the LPS-induced formation of IL-1β. If enzymes involved in beta-oxidation of fatty acids are important for sufficient responses to LPS is currently not clear. Using cells from various patients with inborn long-chain fatty acid oxidation disorders (lcFAOD), we report that disease-causing deleterious variants of Electron Transfer Flavoprotein Dehydrogenase (ETFDH) and of Very Long Chain Acyl-CoA Dehydrogenase (ACADVL), both cause insufficient inflammatory responses to stimulation with LPS. The insufficiencies included reduced TLR4 expression levels, impaired TLR4 signaling, and reduced or absent induction of pro-inflammatory cytokines such as IL-6. The insufficient responses to LPS were reproduced in cells from healthy controls by targeted loss-of-function of either ETFDH or ACADVL, supporting that the deleterious ETFDH and ACADVL variants cause the attenuated responses to LPS. ETFDH and ACADVL encode two distinct enzymes both involved in fatty acid beta-oxidation, and patients with these deficiencies cannot sufficiently metabolize long-chain fatty acids. We report that genes important for beta-oxidation of long-chain fatty acids are also important for inflammatory responses to an acute immunogen trigger like LPS, which may have important implications for understanding infection and other metabolic stress induced disease pathology in lcFAODs.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
