Yuying Liu, Zeyu Yang, Jie Zhang, Na Guo, Nanxin Liu, Qingqing Zhang, Xintao Dang, Yanchen Li, Jie Zhang and Xiaoyan Pan
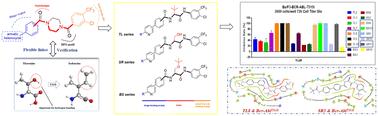
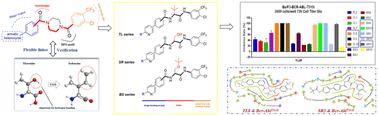
{"title":"Integrating amino acids into Bcr-Abl inhibitors: design, synthesis, biological evaluation, and in silico studies†","authors":"Yuying Liu, Zeyu Yang, Jie Zhang, Na Guo, Nanxin Liu, Qingqing Zhang, Xintao Dang, Yanchen Li, Jie Zhang and Xiaoyan Pan","doi":"10.1039/D4MD00417E","DOIUrl":null,"url":null,"abstract":"<p >Bcr-Abl is successfully applied to drug discovery as a CML therapeutic target, but point mutation resistance has become a major challenge in the clinical treatment of CML. Our previous studies have shown that the introduction of amino acids as flexible linkers and heterocyclic structures as HBMs can achieve potent inhibition of Bcr-Abl<small><sup>T315I</sup></small>. In continuation of these studies, we further enriched the linker types by developing a library of compounds with <em>tert</em>-leucine or serine as a linker. Biological results showed that these compounds exhibited enhanced inhibition against Bcr-Abl<small><sup>WT</sup></small> and Bcr-Abl<small><sup>T315I</sup></small> kinases as well as improved antiproliferative activity in leukemia cell assays compared to previously disclosed compounds. In particular, compounds <strong>TL8</strong>, <strong>TL10</strong>, <strong>BS4</strong>, <strong>BS10</strong>, <strong>SR5</strong> and <strong>SR11</strong> exhibited potent inhibitory activities against Ba/F3 cells bearing a T315I mutant. Additionally, compounds <strong>TL8</strong>, <strong>BS4</strong> and <strong>SR5</strong> effectively induced K562 cell apoptosis, arrested the cell cycle at the S or G2/M phase, and inhibited the phosphorylation of Bcr-Abl and STAT5 in a dose-dependent manner. Docking studies verified the rationality of <em>tert</em>-leucine or serine as a flexible linker and indicated that phenylpyridine with an amide side chain favored the potency of these inhibitors. Moreover, ADME prediction suggested that the tested compounds had a favorable safety profile. Thus, <em>tert</em>-leucine or serine can be used as a promising class of flexible linkers for Bcr-Abl inhibitors with heterocyclic structures as HBMs, and compounds <strong>BS4</strong>, <strong>SR5</strong>, and especially <strong>TL8</strong>, can be used as starting points for further optimization.</p>","PeriodicalId":88,"journal":{"name":"MedChemComm","volume":" 10","pages":" 3507-3528"},"PeriodicalIF":3.5970,"publicationDate":"2024-07-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"MedChemComm","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/md/d4md00417e","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Pharmacology, Toxicology and Pharmaceutics","Score":null,"Total":0}
引用次数: 0
Abstract
Bcr-Abl is successfully applied to drug discovery as a CML therapeutic target, but point mutation resistance has become a major challenge in the clinical treatment of CML. Our previous studies have shown that the introduction of amino acids as flexible linkers and heterocyclic structures as HBMs can achieve potent inhibition of Bcr-AblT315I. In continuation of these studies, we further enriched the linker types by developing a library of compounds with tert-leucine or serine as a linker. Biological results showed that these compounds exhibited enhanced inhibition against Bcr-AblWT and Bcr-AblT315I kinases as well as improved antiproliferative activity in leukemia cell assays compared to previously disclosed compounds. In particular, compounds TL8, TL10, BS4, BS10, SR5 and SR11 exhibited potent inhibitory activities against Ba/F3 cells bearing a T315I mutant. Additionally, compounds TL8, BS4 and SR5 effectively induced K562 cell apoptosis, arrested the cell cycle at the S or G2/M phase, and inhibited the phosphorylation of Bcr-Abl and STAT5 in a dose-dependent manner. Docking studies verified the rationality of tert-leucine or serine as a flexible linker and indicated that phenylpyridine with an amide side chain favored the potency of these inhibitors. Moreover, ADME prediction suggested that the tested compounds had a favorable safety profile. Thus, tert-leucine or serine can be used as a promising class of flexible linkers for Bcr-Abl inhibitors with heterocyclic structures as HBMs, and compounds BS4, SR5, and especially TL8, can be used as starting points for further optimization.
期刊介绍:
Research and review articles in medicinal chemistry and related drug discovery science; the official journal of the European Federation for Medicinal Chemistry.
In 2020, MedChemComm will change its name to RSC Medicinal Chemistry. Issue 12, 2019 will be the last issue as MedChemComm.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
