{"title":"Missense variants of FBN2 associated with congenital arachnodactyly in three Chinese families","authors":"Yu Sui, Yongping Lu, Meina Lin, Xinren Chen, Xiang Ni, Huan Li, Miao Jiang","doi":"10.1016/j.ymgmr.2024.101140","DOIUrl":null,"url":null,"abstract":"<div><h3>Background</h3><p>Congenital contractural arachnodactyly (CCA) is a rare autosomal dominant disorder caused by pathogenic variants of Fibrillin-2 (<em>FBN2</em>) gene. This study aimed to investigate the variants in three Chinese families with CCA.</p></div><div><h3>Methods</h3><p>Next-generation sequencing analysis and Sanger sequencing of exons 24–35 of <em>FBN2</em> (NM_001999.4) were performed on the three CCA pedigrees. The pathogenicity of the variants was assessed using ACMG criteria and predicted using an in-silico program.</p></div><div><h3>Results</h3><p>A novel heterozygous substitution (NM_001999.4: c.3230G > A; NP_001990.2 p. Cys1077Tyr) was identified in all patients from pedigree A, but not in healthy family members. The variant was found to be pathogenic. Additionally, in pedigree B (NM_001999.4: c.4222G > A; NP_001990.2: p.Asp1408Asn) and C (NM_001999.4: c.3170G > A; NP_001990.2: p.Gly1057Asp), and the previously reported variants were detected. Variants affecting cysteine residues may disrupt disulfide bridging, leading to a weakened microfibril scaffold, resulting in CCA phenotypes. High phenotypic heterogeneity was observed among different families, and there was little correlation between the genotype and phenotype.</p></div><div><h3>Conclusion</h3><p>This study describes three large families with CCA caused by missense variants in the <em>FBN2</em> gene. Phenotypic variations were observed among different pedigree groups, and further research is needed to investigate the underlying reasons for these variations.</p></div>","PeriodicalId":18814,"journal":{"name":"Molecular Genetics and Metabolism Reports","volume":"41 ","pages":"Article 101140"},"PeriodicalIF":1.9000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2214426924000934/pdfft?md5=65ae1d4fa45d800bf053f549131f8b0e&pid=1-s2.0-S2214426924000934-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics and Metabolism Reports","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2214426924000934","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/9 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background
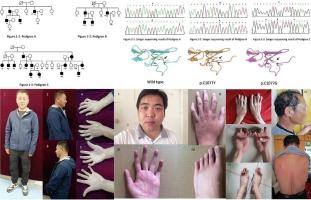
Congenital contractural arachnodactyly (CCA) is a rare autosomal dominant disorder caused by pathogenic variants of Fibrillin-2 (FBN2) gene. This study aimed to investigate the variants in three Chinese families with CCA.
Methods
Next-generation sequencing analysis and Sanger sequencing of exons 24–35 of FBN2 (NM_001999.4) were performed on the three CCA pedigrees. The pathogenicity of the variants was assessed using ACMG criteria and predicted using an in-silico program.
Results
A novel heterozygous substitution (NM_001999.4: c.3230G > A; NP_001990.2 p. Cys1077Tyr) was identified in all patients from pedigree A, but not in healthy family members. The variant was found to be pathogenic. Additionally, in pedigree B (NM_001999.4: c.4222G > A; NP_001990.2: p.Asp1408Asn) and C (NM_001999.4: c.3170G > A; NP_001990.2: p.Gly1057Asp), and the previously reported variants were detected. Variants affecting cysteine residues may disrupt disulfide bridging, leading to a weakened microfibril scaffold, resulting in CCA phenotypes. High phenotypic heterogeneity was observed among different families, and there was little correlation between the genotype and phenotype.
Conclusion
This study describes three large families with CCA caused by missense variants in the FBN2 gene. Phenotypic variations were observed among different pedigree groups, and further research is needed to investigate the underlying reasons for these variations.
期刊介绍:
Molecular Genetics and Metabolism Reports is an open access journal that publishes molecular and metabolic reports describing investigations that use the tools of biochemistry and molecular biology for studies of normal and diseased states. In addition to original research articles, sequence reports, brief communication reports and letters to the editor are considered.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
