{"title":"Deciphering transcriptional bursts and enhancer dynamics: Advancing cancer therapeutics through single-cell global run-on sequencing","authors":"Xiangyu Pan, Feifei Na, Xuelan Chen","doi":"10.1002/mog2.88","DOIUrl":null,"url":null,"abstract":"<p>The pioneering work of Phillip A. Sharp's research group has led to the development of a cutting-edge single-cell nascent RNA sequencing assay, revolutionizing our understanding of transcription dynamics. The study, published in Nature, incorporates click chemistry into global run-on and sequencing (GRO-seq) to create a single-cell GRO-seq (scGRO-seq) technique.<span><sup>1</sup></span> This method allows for the precise capture of the episodic and coordinated nature of transcription at high resolution, revealing critical dynamics such as burst size and enhancer-gene interactions. Such insights are particularly vital for unraveling the complexities of transcription regulation and cell cycle dynamics across various developmental stages and in the pathological context of diseases like cancer.</p><p>Transcription in development and cancer biology involves short bursts of activity and lengthy silent periods, essential for gene regulation. Core regulatory elements like promoters, transcription factors, and enhancers play key roles in these bursts. Enhancers, specific to cell types and states, regulate genes over long distances and are often linked to disease regions, making them potential targets for cancer therapies.<span><sup>2</sup></span> Current genomic tools provide insights into gene activation precursors but lack real-time transcription event capture. scGRO-seq addresses this gap, offering a dynamic view of regulatory mechanisms for targeted cancer treatment.</p><p>The scGRO-seq technique offers a novel and advanced method for analyzing genome-wide transcription dynamics at a single-cell resolution, filling critical gaps in understanding transcriptional regulation. This method involves labeling nascent RNAs with modified nucleotide triphosphates that contain alkyne groups during a nuclear run-on reaction (Figure 1A). These RNAs are subsequently linked to azide-tagged, single-cell barcode DNA molecules through copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC), known for its efficiency and robustness.<span><sup>3</sup></span> This process not only preserves the integrity of the nuclear membrane but also selectively enriches nascent RNA. Postreaction, these barcoded RNAs are pooled, reverse-transcribed, and PCR amplified to construct a sequencing library, which is then used to detail transcriptional activity within individual cells (Figure 1A).</p><p>The scGRO-seq method has revolutionized the understanding of transcriptional dynamics by refining the approach to detect transcription bursts. Focusing on a 10 kb central gene region and excluding the ends with paused polymerases, it utilizes an RNA Polymerase II elongation rate of 2.5 kb/min, limiting the burst detection window to just 4 min. This setup enables precise measurement of burst sizes, which range from 1 to 4 RNA polymerases, with an average of 1.23, and a mean interval between bursts that aligns with the previously reported 2-h global nascent transcription cycle by intron seqFISH. Simulations validating this approach have shown strong correlations with intron seqFISH for genes with high burst frequencies, although correlations with scRNA-seq were less robust, indicating limitations in deriving kinetic estimates from mature transcripts. This innovative method offers a comprehensive and accurate map of transcription activity within individual cells, significantly enhancing our understanding of cellular transcriptional dynamics.</p><p>Further insights from scGRO-seq include its ability to identify nonpolyadenylated, replication-dependent histone genes active exclusively during the S phase of the cell cycle, a feature often missed by traditional scRNA-seq due to the lack of polyadenylation. This capability allows for accurate classification of cell cycle phases and the observation of transcriptional changes during critical cycle transitions, such as DNA replication and recovery phases (Figure 1B). Additionally, scGRO-seq has challenged previous notions about gene co-transcription by providing detailed insights into the transcriptional coordination of functionally related genes. Through stringent analysis within a 4-min window, it was found that only 0.7% of over 112 million gene pairs tested were significantly co-transcribed, forming a network of 59 distinct modules with roles in cell cycle regulation, RNA splicing, and DNA repair. This suggests a sophisticated interplay of transcriptional regulation that could be influenced by common transcription factors or mechanistic gene couplings across different chromosome regions, shedding new light on the complexities of gene expression regulation across vital cellular processes.</p><p>scGRO-seq has revolutionized the understanding of enhancer-gene interactions by precisely capturing and analyzing transcripts from both genes and active enhancers within single cells. This technique focuses on active transcription areas by excluding regions known for transcriptional pausing and analyzing scGRO-seq reads across selected genomic regions. Detailed permutation and correlation analyses of nearly 7 million enhancer-gene pairs identified a significant, though small, subset (0.6%) that exhibited co-transcription within 200 kb. Notably, super-enhancers showed even stronger correlations with gene transcription up to 400 kb away, highlighting a spatial component essential for cell cycle regulation. Further exploration of the temporal dynamics revealed that enhancer activity often precedes transcription at associated gene promoters, suggesting a potential initiating role for enhancers in gene expression. This phenomenon was particularly evident in the transcription of pluripotency-related genes like Sox2 and Nanog4,<span><sup>4</sup></span> where enhancers were active before the genes themselves (Figure 1C). More granular analysis using smaller genomic bins confirmed that enhancer activity could indeed initiate transcription across multiple enhancer-gene pairs. These insights, supported by CRISPR perturbation studies showing decreased gene expression following enhancer disruption, underscore a complex and finely tuned temporal regulation mechanism. This advanced understanding is crucial for comprehending the regulatory landscape governing gene expression, both in the context of embryo development and cancer cell dynamics, and for potential therapeutic interventions.</p><p>The challenges of targeting transcriptional enhancers in cancer therapy are underscored by the complex interactions between enhancers and genes, and emergence of resistance mechanisms. For instance, the use of Bromodomain and Extra-Terminal domain (BET) inhibitors in prostate cancer and leukemia can inadvertently activate alternative pathways such as CDK9 and Wnt signaling, which contribute to resistance by affecting gene expression and cancer cell survival (Figure 1D). This interconnectedness highlights the need for a multifaceted treatment strategy addressing multiple regulatory pathways to effectively manage or prevent resistance. Furthermore, the effectiveness of enhancer-targeting therapies varies across cancer types, largely due to specific enhancer-gene configurations and genetic contexts. For example, EZH2 inhibitors are beneficial in lymphomas characterized by abnormal histone methylation but can worsen conditions such as diffuse intrinsic pontine gliomas and neurofibromatosis, which have different genetic alterations (Figure 1D). Similarly, while histone deacetylase inhibitors are FDA-approved for certain lymphomas and myelomas, their efficacy is limited in other cancers unless combined with therapies like BET inhibitors or chemoradiation<span><sup>5</sup></span> (Figure 1D). These observations emphasize the need for precisely targeted approaches and a deeper understanding of enhancer programming in developing cancer treatments. The future implications of scGRO-seq in this context are profound. By providing high-resolution, single-cell insights into enhancer-gene interactions and transcriptional dynamics, scGRO-seq can identify novel enhancer targets and regulatory networks specific to different cancer types. This technique can reveal how specific enhancers contribute to drug resistance and aid in designing combination therapies pre-emptively target compensatory pathways. Additionally, scGRO-seq allows detailed mapping of enhancer-gene dynamics facilitates, the development of drugs that specifically modulate enhancer activities by inhibiting enhancer functions or altering transcriptional machinery in tumorigenesis.</p><p>By detecting co-transcription within exact temporal windows, scGRO-seq offers insights into how enhancers directly impact gene activation, deepening our understanding of gene regulation in developmental and cancerous cells (Figure 1C,D). Identifying enhancer-gene pairs associated with poor prognosis or resistance to therapy could lead to the development of targeted treatments, such as small molecule inhibitors or antisense oligonucleotides. Integrating these targeted therapies with standard treatments could enhance their efficacy and combat resistance, ushering in a new era of precision oncology focused on the regulatory architecture of cancer cells.</p><p>All authors were involved in the writing of the manuscript. Xiangyu Pan and Xuelan Chen initiated the conception and outline. Xiangyu Pan organized and processed the figure. Xiangyu Pan and Feifei Na revised the manuscript. Xuelan Chen and Feifei Na were involved in study supervision. All authors have read and approved the final manuscript.</p><p>The authors declare no conflict of interest.</p><p>Not applicable.</p>","PeriodicalId":100902,"journal":{"name":"MedComm – Oncology","volume":"3 3","pages":""},"PeriodicalIF":2.2000,"publicationDate":"2024-09-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/mog2.88","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"MedComm – Oncology","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/mog2.88","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
The pioneering work of Phillip A. Sharp's research group has led to the development of a cutting-edge single-cell nascent RNA sequencing assay, revolutionizing our understanding of transcription dynamics. The study, published in Nature, incorporates click chemistry into global run-on and sequencing (GRO-seq) to create a single-cell GRO-seq (scGRO-seq) technique.1 This method allows for the precise capture of the episodic and coordinated nature of transcription at high resolution, revealing critical dynamics such as burst size and enhancer-gene interactions. Such insights are particularly vital for unraveling the complexities of transcription regulation and cell cycle dynamics across various developmental stages and in the pathological context of diseases like cancer.
Transcription in development and cancer biology involves short bursts of activity and lengthy silent periods, essential for gene regulation. Core regulatory elements like promoters, transcription factors, and enhancers play key roles in these bursts. Enhancers, specific to cell types and states, regulate genes over long distances and are often linked to disease regions, making them potential targets for cancer therapies.2 Current genomic tools provide insights into gene activation precursors but lack real-time transcription event capture. scGRO-seq addresses this gap, offering a dynamic view of regulatory mechanisms for targeted cancer treatment.
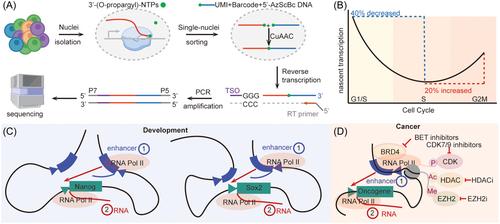
The scGRO-seq technique offers a novel and advanced method for analyzing genome-wide transcription dynamics at a single-cell resolution, filling critical gaps in understanding transcriptional regulation. This method involves labeling nascent RNAs with modified nucleotide triphosphates that contain alkyne groups during a nuclear run-on reaction (Figure 1A). These RNAs are subsequently linked to azide-tagged, single-cell barcode DNA molecules through copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC), known for its efficiency and robustness.3 This process not only preserves the integrity of the nuclear membrane but also selectively enriches nascent RNA. Postreaction, these barcoded RNAs are pooled, reverse-transcribed, and PCR amplified to construct a sequencing library, which is then used to detail transcriptional activity within individual cells (Figure 1A).
The scGRO-seq method has revolutionized the understanding of transcriptional dynamics by refining the approach to detect transcription bursts. Focusing on a 10 kb central gene region and excluding the ends with paused polymerases, it utilizes an RNA Polymerase II elongation rate of 2.5 kb/min, limiting the burst detection window to just 4 min. This setup enables precise measurement of burst sizes, which range from 1 to 4 RNA polymerases, with an average of 1.23, and a mean interval between bursts that aligns with the previously reported 2-h global nascent transcription cycle by intron seqFISH. Simulations validating this approach have shown strong correlations with intron seqFISH for genes with high burst frequencies, although correlations with scRNA-seq were less robust, indicating limitations in deriving kinetic estimates from mature transcripts. This innovative method offers a comprehensive and accurate map of transcription activity within individual cells, significantly enhancing our understanding of cellular transcriptional dynamics.
Further insights from scGRO-seq include its ability to identify nonpolyadenylated, replication-dependent histone genes active exclusively during the S phase of the cell cycle, a feature often missed by traditional scRNA-seq due to the lack of polyadenylation. This capability allows for accurate classification of cell cycle phases and the observation of transcriptional changes during critical cycle transitions, such as DNA replication and recovery phases (Figure 1B). Additionally, scGRO-seq has challenged previous notions about gene co-transcription by providing detailed insights into the transcriptional coordination of functionally related genes. Through stringent analysis within a 4-min window, it was found that only 0.7% of over 112 million gene pairs tested were significantly co-transcribed, forming a network of 59 distinct modules with roles in cell cycle regulation, RNA splicing, and DNA repair. This suggests a sophisticated interplay of transcriptional regulation that could be influenced by common transcription factors or mechanistic gene couplings across different chromosome regions, shedding new light on the complexities of gene expression regulation across vital cellular processes.
scGRO-seq has revolutionized the understanding of enhancer-gene interactions by precisely capturing and analyzing transcripts from both genes and active enhancers within single cells. This technique focuses on active transcription areas by excluding regions known for transcriptional pausing and analyzing scGRO-seq reads across selected genomic regions. Detailed permutation and correlation analyses of nearly 7 million enhancer-gene pairs identified a significant, though small, subset (0.6%) that exhibited co-transcription within 200 kb. Notably, super-enhancers showed even stronger correlations with gene transcription up to 400 kb away, highlighting a spatial component essential for cell cycle regulation. Further exploration of the temporal dynamics revealed that enhancer activity often precedes transcription at associated gene promoters, suggesting a potential initiating role for enhancers in gene expression. This phenomenon was particularly evident in the transcription of pluripotency-related genes like Sox2 and Nanog4,4 where enhancers were active before the genes themselves (Figure 1C). More granular analysis using smaller genomic bins confirmed that enhancer activity could indeed initiate transcription across multiple enhancer-gene pairs. These insights, supported by CRISPR perturbation studies showing decreased gene expression following enhancer disruption, underscore a complex and finely tuned temporal regulation mechanism. This advanced understanding is crucial for comprehending the regulatory landscape governing gene expression, both in the context of embryo development and cancer cell dynamics, and for potential therapeutic interventions.
The challenges of targeting transcriptional enhancers in cancer therapy are underscored by the complex interactions between enhancers and genes, and emergence of resistance mechanisms. For instance, the use of Bromodomain and Extra-Terminal domain (BET) inhibitors in prostate cancer and leukemia can inadvertently activate alternative pathways such as CDK9 and Wnt signaling, which contribute to resistance by affecting gene expression and cancer cell survival (Figure 1D). This interconnectedness highlights the need for a multifaceted treatment strategy addressing multiple regulatory pathways to effectively manage or prevent resistance. Furthermore, the effectiveness of enhancer-targeting therapies varies across cancer types, largely due to specific enhancer-gene configurations and genetic contexts. For example, EZH2 inhibitors are beneficial in lymphomas characterized by abnormal histone methylation but can worsen conditions such as diffuse intrinsic pontine gliomas and neurofibromatosis, which have different genetic alterations (Figure 1D). Similarly, while histone deacetylase inhibitors are FDA-approved for certain lymphomas and myelomas, their efficacy is limited in other cancers unless combined with therapies like BET inhibitors or chemoradiation5 (Figure 1D). These observations emphasize the need for precisely targeted approaches and a deeper understanding of enhancer programming in developing cancer treatments. The future implications of scGRO-seq in this context are profound. By providing high-resolution, single-cell insights into enhancer-gene interactions and transcriptional dynamics, scGRO-seq can identify novel enhancer targets and regulatory networks specific to different cancer types. This technique can reveal how specific enhancers contribute to drug resistance and aid in designing combination therapies pre-emptively target compensatory pathways. Additionally, scGRO-seq allows detailed mapping of enhancer-gene dynamics facilitates, the development of drugs that specifically modulate enhancer activities by inhibiting enhancer functions or altering transcriptional machinery in tumorigenesis.
By detecting co-transcription within exact temporal windows, scGRO-seq offers insights into how enhancers directly impact gene activation, deepening our understanding of gene regulation in developmental and cancerous cells (Figure 1C,D). Identifying enhancer-gene pairs associated with poor prognosis or resistance to therapy could lead to the development of targeted treatments, such as small molecule inhibitors or antisense oligonucleotides. Integrating these targeted therapies with standard treatments could enhance their efficacy and combat resistance, ushering in a new era of precision oncology focused on the regulatory architecture of cancer cells.
All authors were involved in the writing of the manuscript. Xiangyu Pan and Xuelan Chen initiated the conception and outline. Xiangyu Pan organized and processed the figure. Xiangyu Pan and Feifei Na revised the manuscript. Xuelan Chen and Feifei Na were involved in study supervision. All authors have read and approved the final manuscript.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
