Irina D. Yushina, Artëm E. Masunov, Ekaterina V. Bartashevich
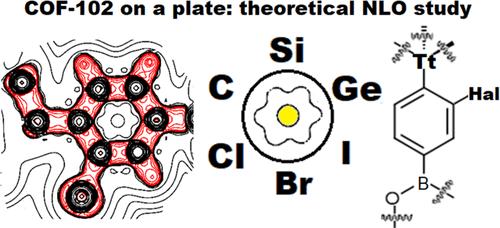
{"title":"Covalent Organic Frameworks in Computational Design of Second-Harmonic Generation Materials: Role of Tetrel Atoms and Their Interactions","authors":"Irina D. Yushina, Artëm E. Masunov, Ekaterina V. Bartashevich","doi":"10.1021/acs.jpca.4c04633","DOIUrl":null,"url":null,"abstract":"Modern approaches to the design of nonlinear optical materials often rely on computational techniques. Here, we discuss the effects of the variation in the center tetrel atoms, Tt = C, Si, or Ge, in a series of covalent organic frameworks of the COF-102 family. The effects of halogen substitution, Hal = Cl, Br, or I on intramolecular tetrel bonding are also discussed. The characteristics of the calculated electron density have been implemented to describe the features of the electron distribution around the central fragment involving a tetrahedral tetrel atom. The effect of the central Tt atom leads to a dramatic change in the character of electron delocalization on the Tt–C<sub>ar</sub> bond with aromatic rings. The location of the halogen atom at the <i>ortho</i>-position of the aromatic ring leads to the formation of tetrel bonds, halogen bonds, or other noncovalent interactions. The changes in the second-order electric susceptibility χ(2) have been studied in order to describe the strength of nonlinear optical properties within the periodic couple-perturbed Kohn–Sham approach. A counterintuitive trend for the χ(2) decrease is observed upon substitution of H > Cl > Br > I at the <i>ortho</i>-position of the phenyl ring. This is due to the corresponding elongation of the Tt–C<sub>ar</sub> bond.","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"108 1","pages":""},"PeriodicalIF":2.8000,"publicationDate":"2024-09-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpca.4c04633","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
Modern approaches to the design of nonlinear optical materials often rely on computational techniques. Here, we discuss the effects of the variation in the center tetrel atoms, Tt = C, Si, or Ge, in a series of covalent organic frameworks of the COF-102 family. The effects of halogen substitution, Hal = Cl, Br, or I on intramolecular tetrel bonding are also discussed. The characteristics of the calculated electron density have been implemented to describe the features of the electron distribution around the central fragment involving a tetrahedral tetrel atom. The effect of the central Tt atom leads to a dramatic change in the character of electron delocalization on the Tt–Car bond with aromatic rings. The location of the halogen atom at the ortho-position of the aromatic ring leads to the formation of tetrel bonds, halogen bonds, or other noncovalent interactions. The changes in the second-order electric susceptibility χ(2) have been studied in order to describe the strength of nonlinear optical properties within the periodic couple-perturbed Kohn–Sham approach. A counterintuitive trend for the χ(2) decrease is observed upon substitution of H > Cl > Br > I at the ortho-position of the phenyl ring. This is due to the corresponding elongation of the Tt–Car bond.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
