Jiachen Zhu, Wei Hong, Tingyu Liu, Hao Hu and Longfeng Zhao
{"title":"First-principles investigation of reduced KDP crystal damage threshold: defect clusters in Mg-related configurations","authors":"Jiachen Zhu, Wei Hong, Tingyu Liu, Hao Hu and Longfeng Zhao","doi":"10.1039/D4CE00624K","DOIUrl":null,"url":null,"abstract":"<p >In this study, we utilized first-principles methods to delve into defect clusters within potassium dihydrogen phosphate (KDP) crystals, focusing on (Mg<small><sub>K</sub></small> + V<small><sub>K</sub></small>) and (Mg<small><sub>K</sub></small> + V<small><sub>H</sub></small>) configurations. We examined their stability, defect formation energy, lattice distortion, electronic structures, and optical properties in both paraelectric (PE-KDP) and ferroelectric (FE-KDP) phases. In the PE phase, compensation of <img> was accomplished <em>via</em> the nearest neighbor <img>. Conversely, in the FE phase, compensation of <img> was achieved utilizing the next nearest neighbor <img>. Notably, the Mg–O ionic bond displayed significant changes in bond length, with a maximum alteration of 60%, as neighboring oxygen atoms moved closer to the magnesium atom. Furthermore, both structures displayed a downward shift of the conduction band minimum (CBM), primarily due to contributions from Mg 3s and O 2p orbitals, resulting in a reduction in the band gap. By analyzing the photoluminescence process alongside electron–phonon coupling phenomena, absorption and emission spectra were obtained. In the absorption spectra, peaks for PE-KDP and FE-KDP were observed at 335 nm and 386 nm, respectively, consistent with experimental observations of absorption at 355 nm. Upon exposure to a 355 nm laser, local crystal absorption led to a progressive increase in temperature, consequently lowering the damage threshold.</p>","PeriodicalId":70,"journal":{"name":"CrystEngComm","volume":" 37","pages":" 5267-5277"},"PeriodicalIF":2.6000,"publicationDate":"2024-09-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"CrystEngComm","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/ce/d4ce00624k","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
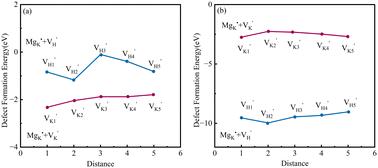
In this study, we utilized first-principles methods to delve into defect clusters within potassium dihydrogen phosphate (KDP) crystals, focusing on (MgK + VK) and (MgK + VH) configurations. We examined their stability, defect formation energy, lattice distortion, electronic structures, and optical properties in both paraelectric (PE-KDP) and ferroelectric (FE-KDP) phases. In the PE phase, compensation of was accomplished via the nearest neighbor . Conversely, in the FE phase, compensation of was achieved utilizing the next nearest neighbor . Notably, the Mg–O ionic bond displayed significant changes in bond length, with a maximum alteration of 60%, as neighboring oxygen atoms moved closer to the magnesium atom. Furthermore, both structures displayed a downward shift of the conduction band minimum (CBM), primarily due to contributions from Mg 3s and O 2p orbitals, resulting in a reduction in the band gap. By analyzing the photoluminescence process alongside electron–phonon coupling phenomena, absorption and emission spectra were obtained. In the absorption spectra, peaks for PE-KDP and FE-KDP were observed at 335 nm and 386 nm, respectively, consistent with experimental observations of absorption at 355 nm. Upon exposure to a 355 nm laser, local crystal absorption led to a progressive increase in temperature, consequently lowering the damage threshold.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
