{"title":"Ab Initio Calculations of the Electronic Structure of the Doublet and Quartet States of the Rubidium Trimer","authors":"E. A. Bormotova, A. S. Likharev, A. V. Stolyarov","doi":"10.1134/S0030400X24030044","DOIUrl":null,"url":null,"abstract":"<p>Systematic quantum chemical calculations were performed for the ground and a number of low-lying electronically excited doublet and quartet states of the rubidium trimer molecule. The obtained potential energy surfaces (PES), spin-orbit couplings (SOC) and electronic transition dipole moments (ETDM) can be useful for optimizing paths for laser synthesis, cooling and manipulation of stable ensembles of Rb<sub>3</sub> molecules at ultralow temperatures. Ab initio calculations of the electronic structure of the homonuclear Rb<sub>3</sub> molecule, in linear, isosceles triangle and equilateral triangle geometries, were performed using the multi-reference configuration interaction method, taking into account single and double excitations (M-R‑CISD) and with explicit dynamic correlation of only the three valence electrons. The structure of each atom was approximated using a nine-electron effective core potential (ECP28MDF), and molecular orbitals (MOs) were optimized using the spin averaged (over doublet and quartet states) multi-configuration self-consistent field (SA-CASSCF) method. Core–valence correlations between twenty-four subvalence electrons located on doubly occupied MOs and three valence electrons were implicitly taken into account using a one-electron angular momentum-independent Müller–Mayer core polarization potential (CPP). As a result of topological investigations at over 35 000 points, two dimensional PES, SOC, and ETDM functions were obtained and the geometric parameters Rb<sub>3</sub> were found at which the most intense vertical transitions and the maximum influence of the SOC are expected.</p>","PeriodicalId":723,"journal":{"name":"Optics and Spectroscopy","volume":"132 3","pages":"223 - 233"},"PeriodicalIF":0.9000,"publicationDate":"2024-09-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Optics and Spectroscopy","FirstCategoryId":"101","ListUrlMain":"https://link.springer.com/article/10.1134/S0030400X24030044","RegionNum":4,"RegionCategory":"物理与天体物理","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"OPTICS","Score":null,"Total":0}
引用次数: 0
Abstract
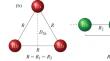
Systematic quantum chemical calculations were performed for the ground and a number of low-lying electronically excited doublet and quartet states of the rubidium trimer molecule. The obtained potential energy surfaces (PES), spin-orbit couplings (SOC) and electronic transition dipole moments (ETDM) can be useful for optimizing paths for laser synthesis, cooling and manipulation of stable ensembles of Rb3 molecules at ultralow temperatures. Ab initio calculations of the electronic structure of the homonuclear Rb3 molecule, in linear, isosceles triangle and equilateral triangle geometries, were performed using the multi-reference configuration interaction method, taking into account single and double excitations (M-R‑CISD) and with explicit dynamic correlation of only the three valence electrons. The structure of each atom was approximated using a nine-electron effective core potential (ECP28MDF), and molecular orbitals (MOs) were optimized using the spin averaged (over doublet and quartet states) multi-configuration self-consistent field (SA-CASSCF) method. Core–valence correlations between twenty-four subvalence electrons located on doubly occupied MOs and three valence electrons were implicitly taken into account using a one-electron angular momentum-independent Müller–Mayer core polarization potential (CPP). As a result of topological investigations at over 35 000 points, two dimensional PES, SOC, and ETDM functions were obtained and the geometric parameters Rb3 were found at which the most intense vertical transitions and the maximum influence of the SOC are expected.
期刊介绍:
Optics and Spectroscopy (Optika i spektroskopiya), founded in 1956, presents original and review papers in various fields of modern optics and spectroscopy in the entire wavelength range from radio waves to X-rays. Topics covered include problems of theoretical and experimental spectroscopy of atoms, molecules, and condensed state, lasers and the interaction of laser radiation with matter, physical and geometrical optics, holography, and physical principles of optical instrument making.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
