{"title":"Unsupervised Machine Learning in the Analysis of Nonadiabatic Molecular Dynamics Simulation","authors":"Yifei Zhu, Jiawei Peng, Chao Xu, Zhenggang Lan","doi":"10.1021/acs.jpclett.4c01751","DOIUrl":null,"url":null,"abstract":"The all-atomic full-dimensional-level simulations of nonadiabatic molecular dynamics (NAMD) in large realistic systems has received high research interest in recent years. However, such NAMD simulations normally generate an enormous amount of time-dependent high-dimensional data, leading to a significant challenge in result analyses. Based on unsupervised machine learning (ML) methods, considerable efforts were devoted to developing novel and easy-to-use analysis tools for the identification of photoinduced reaction channels and the comprehensive understanding of complicated molecular motions in NAMD simulations. Here, we tried to survey recent advances in this field, particularly to focus on how to use unsupervised ML methods to analyze the trajectory-based NAMD simulation results. Our purpose is to offer a comprehensive discussion on several essential components of this analysis protocol, including the selection of ML methods, the construction of molecular descriptors, the establishment of analytical frameworks, their advantages and limitations, and persistent challenges.","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"8 1","pages":"9601-9619"},"PeriodicalIF":4.6000,"publicationDate":"2024-09-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpclett.4c01751","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
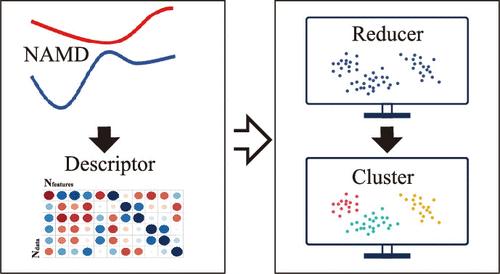
The all-atomic full-dimensional-level simulations of nonadiabatic molecular dynamics (NAMD) in large realistic systems has received high research interest in recent years. However, such NAMD simulations normally generate an enormous amount of time-dependent high-dimensional data, leading to a significant challenge in result analyses. Based on unsupervised machine learning (ML) methods, considerable efforts were devoted to developing novel and easy-to-use analysis tools for the identification of photoinduced reaction channels and the comprehensive understanding of complicated molecular motions in NAMD simulations. Here, we tried to survey recent advances in this field, particularly to focus on how to use unsupervised ML methods to analyze the trajectory-based NAMD simulation results. Our purpose is to offer a comprehensive discussion on several essential components of this analysis protocol, including the selection of ML methods, the construction of molecular descriptors, the establishment of analytical frameworks, their advantages and limitations, and persistent challenges.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
