Reconciling Accuracy and Feasibility for Barrierless Reaction Steps by the PCS/DDCI/MC-PDFT Protocol: Methane and Ethylene Dissociations as Case Studies
{"title":"Reconciling Accuracy and Feasibility for Barrierless Reaction Steps by the PCS/DDCI/MC-PDFT Protocol: Methane and Ethylene Dissociations as Case Studies","authors":"Luigi Crisci, Vincenzo Barone","doi":"10.1021/acs.jctc.4c00911","DOIUrl":null,"url":null,"abstract":"Several enhancements have been introduced into state-of-the-art computational protocols for the treatment of barrierless reaction steps in the framework of variable reaction coordinate variational transition state theory. The first step is the synergistic integration of the Iterative Difference Dedicated Configuration Interaction (I-DDCI) and Pisa Composite Scheme, which defines a reduced cost, yet very accurate, computational workflow. This approach provides a near black box tool for obtaining 1D reference potentials. Then, a general strategy has been devised for tuning the level of theory used in Monte Carlo (MC) sampling, employing Multiconfiguration Pair Density Functional Theory (MC-PDFT) with dynamically adjusted Hartree–Fock exchange. Concurrently, partial geometry optimizations during the MC simulations account for the coupling between the reaction coordinates and conserved modes. The protocol closely approaches full size consistency and yields highly accurate results, with several test computations suggesting rapid convergence of the I-DDCI correction with the basis set dimensions. The capabilities of the new platform are illustrated by two case studies (the hydrogen dissociation from CH<sub>4</sub> and C<sub>2</sub>H<sub>4</sub>), which highlight its flexibility in handling different carbon hybridizations (sp<sup>3</sup> and sp<sup>2</sup>). The remarkable accuracy of the computed rate constants confirms the robustness of the proposed method. Together with their intrinsic interest, these results pave the way for systematic investigations of complex gas-phase reactions through a reliable, user-friendly tool accessible to specialists and nonspecialists alike.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"23 1","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-09-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00911","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
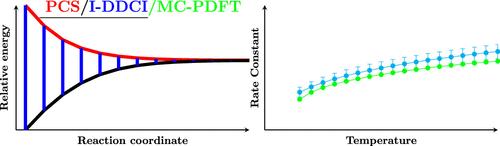
Several enhancements have been introduced into state-of-the-art computational protocols for the treatment of barrierless reaction steps in the framework of variable reaction coordinate variational transition state theory. The first step is the synergistic integration of the Iterative Difference Dedicated Configuration Interaction (I-DDCI) and Pisa Composite Scheme, which defines a reduced cost, yet very accurate, computational workflow. This approach provides a near black box tool for obtaining 1D reference potentials. Then, a general strategy has been devised for tuning the level of theory used in Monte Carlo (MC) sampling, employing Multiconfiguration Pair Density Functional Theory (MC-PDFT) with dynamically adjusted Hartree–Fock exchange. Concurrently, partial geometry optimizations during the MC simulations account for the coupling between the reaction coordinates and conserved modes. The protocol closely approaches full size consistency and yields highly accurate results, with several test computations suggesting rapid convergence of the I-DDCI correction with the basis set dimensions. The capabilities of the new platform are illustrated by two case studies (the hydrogen dissociation from CH4 and C2H4), which highlight its flexibility in handling different carbon hybridizations (sp3 and sp2). The remarkable accuracy of the computed rate constants confirms the robustness of the proposed method. Together with their intrinsic interest, these results pave the way for systematic investigations of complex gas-phase reactions through a reliable, user-friendly tool accessible to specialists and nonspecialists alike.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
