Ian R. Humphreys, Jing Zhang, Minkyung Baek, Yaxi Wang, Aditya Krishnakumar, Jimin Pei, Ivan Anishchenko, Catherine A. Tower, Blake A. Jackson, Thulasi Warrier, Deborah T. Hung, S. Brook Peterson, Joseph D. Mougous, Qian Cong, David Baker
{"title":"Protein interactions in human pathogens revealed through deep learning","authors":"Ian R. Humphreys, Jing Zhang, Minkyung Baek, Yaxi Wang, Aditya Krishnakumar, Jimin Pei, Ivan Anishchenko, Catherine A. Tower, Blake A. Jackson, Thulasi Warrier, Deborah T. Hung, S. Brook Peterson, Joseph D. Mougous, Qian Cong, David Baker","doi":"10.1038/s41564-024-01791-x","DOIUrl":null,"url":null,"abstract":"Identification of bacterial protein–protein interactions and predicting the structures of these complexes could aid in the understanding of pathogenicity mechanisms and developing treatments for infectious diseases. Here we developed RoseTTAFold2-Lite, a rapid deep learning model that leverages residue–residue coevolution and protein structure prediction to systematically identify and structurally characterize protein–protein interactions at the proteome-wide scale. Using this pipeline, we searched through 78 million pairs of proteins across 19 human bacterial pathogens and identified 1,923 confidently predicted complexes involving essential genes and 256 involving virulence factors. Many of these complexes were not previously known; we experimentally tested 12 such predictions, and half of them were validated. The predicted interactions span core metabolic and virulence pathways ranging from post-transcriptional modification to acid neutralization to outer-membrane machinery and should contribute to our understanding of the biology of these important pathogens and the design of drugs to combat them. RoseTTAFold2-Lite uses residue–residue coevolution and protein structure prediction to identify and structurally characterize protein–protein interactions in bacterial pathogens.","PeriodicalId":18992,"journal":{"name":"Nature Microbiology","volume":"9 10","pages":"2642-2652"},"PeriodicalIF":19.4000,"publicationDate":"2024-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s41564-024-01791-x.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Microbiology","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s41564-024-01791-x","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
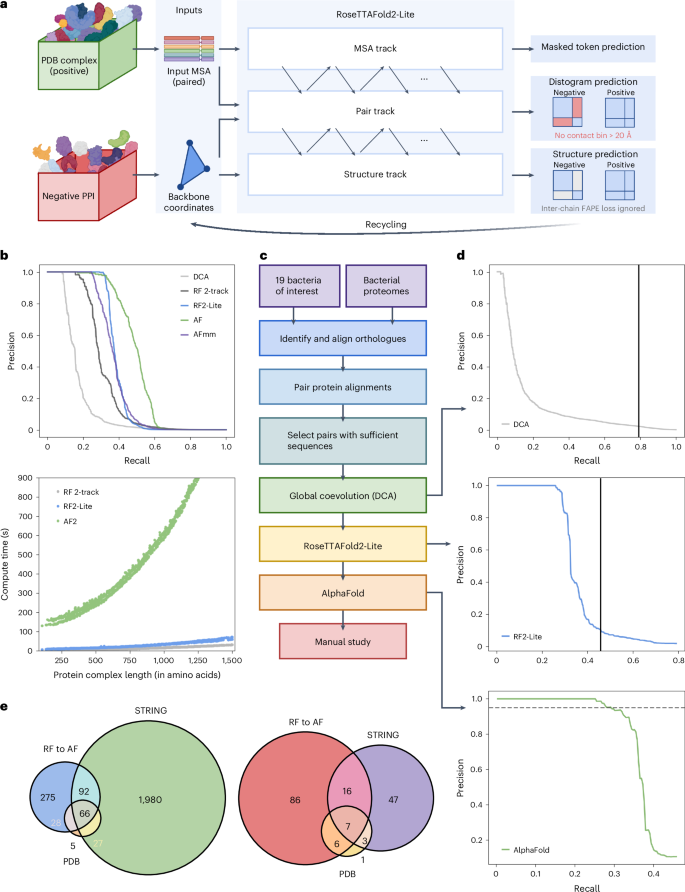
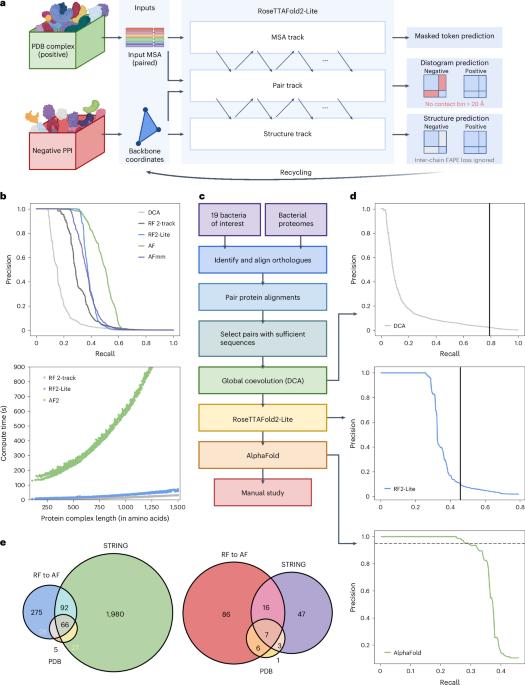
Identification of bacterial protein–protein interactions and predicting the structures of these complexes could aid in the understanding of pathogenicity mechanisms and developing treatments for infectious diseases. Here we developed RoseTTAFold2-Lite, a rapid deep learning model that leverages residue–residue coevolution and protein structure prediction to systematically identify and structurally characterize protein–protein interactions at the proteome-wide scale. Using this pipeline, we searched through 78 million pairs of proteins across 19 human bacterial pathogens and identified 1,923 confidently predicted complexes involving essential genes and 256 involving virulence factors. Many of these complexes were not previously known; we experimentally tested 12 such predictions, and half of them were validated. The predicted interactions span core metabolic and virulence pathways ranging from post-transcriptional modification to acid neutralization to outer-membrane machinery and should contribute to our understanding of the biology of these important pathogens and the design of drugs to combat them. RoseTTAFold2-Lite uses residue–residue coevolution and protein structure prediction to identify and structurally characterize protein–protein interactions in bacterial pathogens.
期刊介绍:
Nature Microbiology aims to cover a comprehensive range of topics related to microorganisms. This includes:
Evolution: The journal is interested in exploring the evolutionary aspects of microorganisms. This may include research on their genetic diversity, adaptation, and speciation over time.
Physiology and cell biology: Nature Microbiology seeks to understand the functions and characteristics of microorganisms at the cellular and physiological levels. This may involve studying their metabolism, growth patterns, and cellular processes.
Interactions: The journal focuses on the interactions microorganisms have with each other, as well as their interactions with hosts or the environment. This encompasses investigations into microbial communities, symbiotic relationships, and microbial responses to different environments.
Societal significance: Nature Microbiology recognizes the societal impact of microorganisms and welcomes studies that explore their practical applications. This may include research on microbial diseases, biotechnology, or environmental remediation.
In summary, Nature Microbiology is interested in research related to the evolution, physiology and cell biology of microorganisms, their interactions, and their societal relevance.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
