Tomasz Bukowy, Matthew L. Brown, Paul L. A. Popelier
{"title":"Toward Gaussian Process Regression Modeling of a Urea Force Field","authors":"Tomasz Bukowy, Matthew L. Brown, Paul L. A. Popelier","doi":"10.1021/acs.jpca.4c04117","DOIUrl":null,"url":null,"abstract":"FFLUX is a next-generation, machine-learnt force field built on three cornerstones: quantum chemical topology, Gaussian process regression, and (high-rank) multipolar electrostatics. It is capable of performing molecular dynamics with near-quantum accuracy at a lower computational cost than standard <i>ab initio</i> molecular dynamics. Previous work with FFLUX was concerned with water and formamide. In this study, we go one step further and challenge FFLUX to model urea, a larger and more flexible system. In result, we have trained urea models at the B3LYP/aug-cc-pVTZ level of theory, with a mean absolute error of 0.4 kJ mol<sup>–1</sup> and a maximum prediction error below 7.0 kJ mol<sup>–1</sup>. To test their performance in molecular dynamics simulations, two sets of FFLUX geometry optimizations were carried out: 5 dimers corresponding to energy minima and 75 random dimers. The 5 dimers were recovered with a root-mean-square deviation below 0.1 Å with respect to their <i>ab initio</i> references. Out of the 75 random dimers, 68% converged to the qualitatively same dimer as those obtained at the <i>ab initio</i> level. Furthermore, we have ranked the 5 FFLUX-optimized dimers in the order of their relative FFLUX single-point energies and compared them with the <i>ab initio</i> method. The energy ranking fully agreed but for one crossover between two successive minima. Finally, we have demonstrated the importance of geometry-dependent (<i>i</i>.<i>e</i>., flexible) multipole moments, showing that the lack of multipole moment flexibility can lead to average errors in the total intermolecular electrostatic energy of more than 2 orders of magnitude.","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"10 1","pages":""},"PeriodicalIF":2.8000,"publicationDate":"2024-09-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpca.4c04117","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
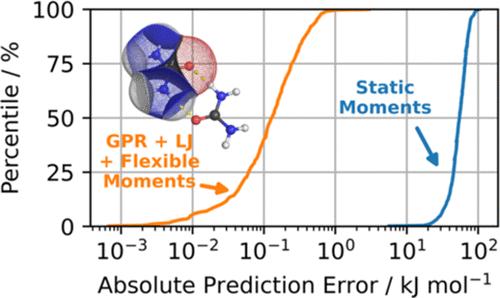
FFLUX is a next-generation, machine-learnt force field built on three cornerstones: quantum chemical topology, Gaussian process regression, and (high-rank) multipolar electrostatics. It is capable of performing molecular dynamics with near-quantum accuracy at a lower computational cost than standard ab initio molecular dynamics. Previous work with FFLUX was concerned with water and formamide. In this study, we go one step further and challenge FFLUX to model urea, a larger and more flexible system. In result, we have trained urea models at the B3LYP/aug-cc-pVTZ level of theory, with a mean absolute error of 0.4 kJ mol–1 and a maximum prediction error below 7.0 kJ mol–1. To test their performance in molecular dynamics simulations, two sets of FFLUX geometry optimizations were carried out: 5 dimers corresponding to energy minima and 75 random dimers. The 5 dimers were recovered with a root-mean-square deviation below 0.1 Å with respect to their ab initio references. Out of the 75 random dimers, 68% converged to the qualitatively same dimer as those obtained at the ab initio level. Furthermore, we have ranked the 5 FFLUX-optimized dimers in the order of their relative FFLUX single-point energies and compared them with the ab initio method. The energy ranking fully agreed but for one crossover between two successive minima. Finally, we have demonstrated the importance of geometry-dependent (i.e., flexible) multipole moments, showing that the lack of multipole moment flexibility can lead to average errors in the total intermolecular electrostatic energy of more than 2 orders of magnitude.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
