Comparative Study of n-C5 Bond Dissociation Energies and H-Atom Abstraction via H for Alkane, Alcohol, Methyl Ether, Nitroso, Nitro, Nitrite, and Nitrate
{"title":"Comparative Study of n-C5 Bond Dissociation Energies and H-Atom Abstraction via H for Alkane, Alcohol, Methyl Ether, Nitroso, Nitro, Nitrite, and Nitrate","authors":"Malte Döntgen, K. Alexander Heufer","doi":"10.1021/acs.jpca.4c05309","DOIUrl":null,"url":null,"abstract":"Energetic materials have a wide range of applications, for many of which new materials are constantly developed. For understanding how energetic materials are chemically converted, it is essential to systematically understand how relevant functional groups affect the stability and properties of such materials. Here, the impact of nitroso, nitro, nitrite, and nitrate functional groups on a pentyl moiety is studied theoretically, focusing on bond dissociation energies and H atom abstraction via Ḣ. The nitroso group is found to imply the strongest effects, while the nitro group appears to exert a stabilizing effect on the pentyl moiety. Importantly, the nitrogenated functional groups generally stabilize the pentyl moiety, in contrast to alcohol and methyl ether functional groups. Therefore, it is concluded that using analogies to the chemistries of the latter two functional groups for chemical kinetic modeling of nitrogenated functional groups is not adequate. Based on the presented bond dissociation energies and rate coefficients, it is to be expected that unimolecular bond fissions at the nitrogenated functional groups will dominate over a wide temperature range. Building on the provided data, future detailed chemical kinetic modeling efforts for nitroso, nitro, nitrite, and nitrate compounds can be aided or initiated.","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"21 1","pages":""},"PeriodicalIF":2.8000,"publicationDate":"2024-09-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpca.4c05309","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
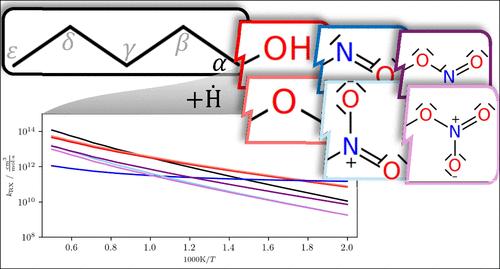
Energetic materials have a wide range of applications, for many of which new materials are constantly developed. For understanding how energetic materials are chemically converted, it is essential to systematically understand how relevant functional groups affect the stability and properties of such materials. Here, the impact of nitroso, nitro, nitrite, and nitrate functional groups on a pentyl moiety is studied theoretically, focusing on bond dissociation energies and H atom abstraction via Ḣ. The nitroso group is found to imply the strongest effects, while the nitro group appears to exert a stabilizing effect on the pentyl moiety. Importantly, the nitrogenated functional groups generally stabilize the pentyl moiety, in contrast to alcohol and methyl ether functional groups. Therefore, it is concluded that using analogies to the chemistries of the latter two functional groups for chemical kinetic modeling of nitrogenated functional groups is not adequate. Based on the presented bond dissociation energies and rate coefficients, it is to be expected that unimolecular bond fissions at the nitrogenated functional groups will dominate over a wide temperature range. Building on the provided data, future detailed chemical kinetic modeling efforts for nitroso, nitro, nitrite, and nitrate compounds can be aided or initiated.
烷烃、酒精、甲醚、亚硝基、硝基、亚硝酸盐和硝酸盐的 n-C5 键解离能和通过 H 原子萃取 H 的比较研究
高能材料具有广泛的应用领域,其中许多领域都在不断开发新材料。要了解高能材料是如何进行化学转化的,就必须系统地了解相关官能团是如何影响这些材料的稳定性和特性的。在此,我们从理论上研究了亚硝基、硝基、亚硝酸盐和硝酸盐官能团对戊基的影响,重点关注键解离能和通过Ḣ抽取 H 原子。研究发现,亚硝基官能团的作用最强,而硝基官能团似乎对戊基分子有稳定作用。重要的是,与醇和甲基醚官能团相比,氮化官能团通常能稳定戊基。因此,可以得出结论,用后两种官能团的化学性质来类比氮化官能团的化学动力学模型是不够的。根据所提供的化学键解离能和速率系数,可以预计在较宽的温度范围内,氮化官能团的单分子化学键裂变将占主导地位。根据所提供的数据,可以帮助或启动未来亚硝基、硝基、亚硝酸盐和硝酸盐化合物的详细化学动力学建模工作。
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
