Omar S. M. El Nahhas, Marko van Treeck, Georg Wölflein, Michaela Unger, Marta Ligero, Tim Lenz, Sophia J. Wagner, Katherine J. Hewitt, Firas Khader, Sebastian Foersch, Daniel Truhn, Jakob Nikolas Kather
{"title":"From whole-slide image to biomarker prediction: end-to-end weakly supervised deep learning in computational pathology","authors":"Omar S. M. El Nahhas, Marko van Treeck, Georg Wölflein, Michaela Unger, Marta Ligero, Tim Lenz, Sophia J. Wagner, Katherine J. Hewitt, Firas Khader, Sebastian Foersch, Daniel Truhn, Jakob Nikolas Kather","doi":"10.1038/s41596-024-01047-2","DOIUrl":null,"url":null,"abstract":"Hematoxylin- and eosin-stained whole-slide images (WSIs) are the foundation of diagnosis of cancer. In recent years, development of deep learning-based methods in computational pathology has enabled the prediction of biomarkers directly from WSIs. However, accurately linking tissue phenotype to biomarkers at scale remains a crucial challenge for democratizing complex biomarkers in precision oncology. This protocol describes a practical workflow for solid tumor associative modeling in pathology (STAMP), enabling prediction of biomarkers directly from WSIs by using deep learning. The STAMP workflow is biomarker agnostic and allows for genetic and clinicopathologic tabular data to be included as an additional input, together with histopathology images. The protocol consists of five main stages that have been successfully applied to various research problems: formal problem definition, data preprocessing, modeling, evaluation and clinical translation. The STAMP workflow differentiates itself through its focus on serving as a collaborative framework that can be used by clinicians and engineers alike for setting up research projects in the field of computational pathology. As an example task, we applied STAMP to the prediction of microsatellite instability (MSI) status in colorectal cancer, showing accurate performance for the identification of tumors high in MSI. Moreover, we provide an open-source code base, which has been deployed at several hospitals across the globe to set up computational pathology workflows. The STAMP workflow requires one workday of hands-on computational execution and basic command line knowledge. We present a practical workflow for end-to-end weakly supervised deep learning to predict biomarkers directly from whole-slide images, enabling clinical researchers to work with engineers to set up a complete computational pathology project.","PeriodicalId":18901,"journal":{"name":"Nature Protocols","volume":"20 1","pages":"293-316"},"PeriodicalIF":16.0000,"publicationDate":"2024-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Protocols","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s41596-024-01047-2","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
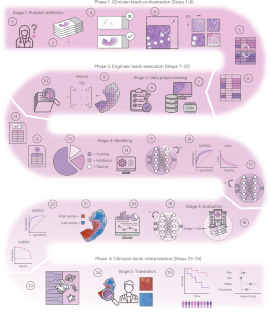
Hematoxylin- and eosin-stained whole-slide images (WSIs) are the foundation of diagnosis of cancer. In recent years, development of deep learning-based methods in computational pathology has enabled the prediction of biomarkers directly from WSIs. However, accurately linking tissue phenotype to biomarkers at scale remains a crucial challenge for democratizing complex biomarkers in precision oncology. This protocol describes a practical workflow for solid tumor associative modeling in pathology (STAMP), enabling prediction of biomarkers directly from WSIs by using deep learning. The STAMP workflow is biomarker agnostic and allows for genetic and clinicopathologic tabular data to be included as an additional input, together with histopathology images. The protocol consists of five main stages that have been successfully applied to various research problems: formal problem definition, data preprocessing, modeling, evaluation and clinical translation. The STAMP workflow differentiates itself through its focus on serving as a collaborative framework that can be used by clinicians and engineers alike for setting up research projects in the field of computational pathology. As an example task, we applied STAMP to the prediction of microsatellite instability (MSI) status in colorectal cancer, showing accurate performance for the identification of tumors high in MSI. Moreover, we provide an open-source code base, which has been deployed at several hospitals across the globe to set up computational pathology workflows. The STAMP workflow requires one workday of hands-on computational execution and basic command line knowledge. We present a practical workflow for end-to-end weakly supervised deep learning to predict biomarkers directly from whole-slide images, enabling clinical researchers to work with engineers to set up a complete computational pathology project.
期刊介绍:
Nature Protocols focuses on publishing protocols used to address significant biological and biomedical science research questions, including methods grounded in physics and chemistry with practical applications to biological problems. The journal caters to a primary audience of research scientists and, as such, exclusively publishes protocols with research applications. Protocols primarily aimed at influencing patient management and treatment decisions are not featured.
The specific techniques covered encompass a wide range, including but not limited to: Biochemistry, Cell biology, Cell culture, Chemical modification, Computational biology, Developmental biology, Epigenomics, Genetic analysis, Genetic modification, Genomics, Imaging, Immunology, Isolation, purification, and separation, Lipidomics, Metabolomics, Microbiology, Model organisms, Nanotechnology, Neuroscience, Nucleic-acid-based molecular biology, Pharmacology, Plant biology, Protein analysis, Proteomics, Spectroscopy, Structural biology, Synthetic chemistry, Tissue culture, Toxicology, and Virology.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
