Feiyun Jia*, Chenghua Zhang, Yongsheng Yang and Bo Zhang*,
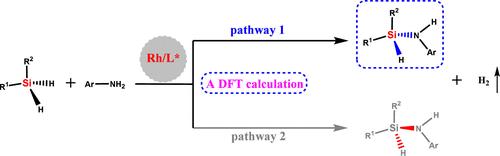
{"title":"Mechanistic Insights into Rh-Catalyzed Asymmetric Synthesis of Silicon-Stereogenic Silazanes: The Origin of Enantioselectivity","authors":"Feiyun Jia*, Chenghua Zhang, Yongsheng Yang and Bo Zhang*, ","doi":"10.1021/acs.organomet.4c0026610.1021/acs.organomet.4c00266","DOIUrl":null,"url":null,"abstract":"<p >The catalytic asymmetric synthesis of silazanes is always a challenging task. Here, a highly enantioselective synthesis of silicon-stereogenic silazanes was investigated to elucidate the protocol’s principal features and to clarify the origin of the enantioselectivity by using DFT calculations. The computational results indicate that the total free energy barrier for the conversion is 19.9 kcal/mol, which is reasonable given the current reaction conditions. Consistent with the experimental findings, the calculations indicate that σ-bond metathesis (N–H bond cleavage) is the rate-determining step for this transformation. Both pathways 1 and 2 toward <i>S</i>- or <i>R</i>-configuration products were investigated computationally. We found that the main enantiomer product of this transformation is determined by the kinetically more favorable main reaction pathway 1. Calculations indicate that the loss of one or the other H on the dihydrosilane will lock the product chirality; therefore, the oxidative addition is the enantioselectivity-determining step. Non-covalent interaction (NCI) analysis confirms that a difference in steric hindrance is responsible for the enantioselectivity of the protocol. Additionally, calculations confirm that the electron-donating group on aniline appropriately lowers the free energy barrier relative to the electron-withdrawing group (Δ<i>G</i> = 15.5 vs 21.6 kcal/mol), thereby accelerating the conversion.</p>","PeriodicalId":56,"journal":{"name":"Organometallics","volume":null,"pages":null},"PeriodicalIF":2.5000,"publicationDate":"2024-09-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Organometallics","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.organomet.4c00266","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
引用次数: 0
Abstract
The catalytic asymmetric synthesis of silazanes is always a challenging task. Here, a highly enantioselective synthesis of silicon-stereogenic silazanes was investigated to elucidate the protocol’s principal features and to clarify the origin of the enantioselectivity by using DFT calculations. The computational results indicate that the total free energy barrier for the conversion is 19.9 kcal/mol, which is reasonable given the current reaction conditions. Consistent with the experimental findings, the calculations indicate that σ-bond metathesis (N–H bond cleavage) is the rate-determining step for this transformation. Both pathways 1 and 2 toward S- or R-configuration products were investigated computationally. We found that the main enantiomer product of this transformation is determined by the kinetically more favorable main reaction pathway 1. Calculations indicate that the loss of one or the other H on the dihydrosilane will lock the product chirality; therefore, the oxidative addition is the enantioselectivity-determining step. Non-covalent interaction (NCI) analysis confirms that a difference in steric hindrance is responsible for the enantioselectivity of the protocol. Additionally, calculations confirm that the electron-donating group on aniline appropriately lowers the free energy barrier relative to the electron-withdrawing group (ΔG = 15.5 vs 21.6 kcal/mol), thereby accelerating the conversion.
期刊介绍:
Organometallics is the flagship journal of organometallic chemistry and records progress in one of the most active fields of science, bridging organic and inorganic chemistry. The journal publishes Articles, Communications, Reviews, and Tutorials (instructional overviews) that depict research on the synthesis, structure, bonding, chemical reactivity, and reaction mechanisms for a variety of applications, including catalyst design and catalytic processes; main-group, transition-metal, and lanthanide and actinide metal chemistry; synthetic aspects of polymer science and materials science; and bioorganometallic chemistry.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
