A Computational Study of Gold(I)-Catalyzed Isomerization of Cyclooctyne: A Case Study on the Mechanism of C(sp3)–H Insertion by Cationic Gold Alkyne Complexes and Model Studies
Harrison E. Bruggeman, Rachel Lorson, Lilia J. Allen, Logan G. Jackson, Winston Gee and Brandon E. Haines*,
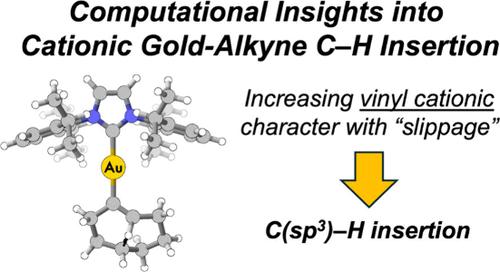
{"title":"A Computational Study of Gold(I)-Catalyzed Isomerization of Cyclooctyne: A Case Study on the Mechanism of C(sp3)–H Insertion by Cationic Gold Alkyne Complexes and Model Studies","authors":"Harrison E. Bruggeman, Rachel Lorson, Lilia J. Allen, Logan G. Jackson, Winston Gee and Brandon E. Haines*, ","doi":"10.1021/acs.organomet.4c0035910.1021/acs.organomet.4c00359","DOIUrl":null,"url":null,"abstract":"<p >Cationic gold-alkyne chemistry is a vital component of homogeneous gold catalysis due to its ability to access a wide range of reactive intermediates. Direct C(sp<sup>3</sup>)–H insertion by the cationic gold-alkyne complex is an emergent reaction in this area without a well-defined reactive intermediate. Gold(I)-catalyzed isomerization of cyclooctyne facilitated by <i>trans</i>-annular C(sp<sup>3</sup>)–H insertion ( <cite><i>Eur. J. Inorg. Chem.</i></cite> <span>2016</span>, 995−1001) is used as a case study to investigate the mechanism of this process with density functional theory (DFT) calculations. Natural resonance theory (NRT) calculations are used to analyze the reactive intermediate in terms of familiar resonance structures. It is found that “slippage” or deformation of the gold ion coordination from η<sup>2</sup> to η<sup>1</sup> increases the NRT weighting of the vinyl cation resonance structure from 12% to 25% leading to its C(sp<sup>3</sup>)–H insertion reactivity. In addition, DFT calculations and a distortion-interaction analysis are used to rationalize the catalyst-dependent regioselectivity observed in the reaction. Lastly, model studies investigating the impact of the alkyne substrate and ancillary ligand show that electron-withdrawing substituents and electron deficient ligands lower Gibbs activation energy for C(sp<sup>3</sup>)–H insertion, which suggests strategies to further improve the reaction through catalyst design.</p>","PeriodicalId":56,"journal":{"name":"Organometallics","volume":null,"pages":null},"PeriodicalIF":2.5000,"publicationDate":"2024-09-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Organometallics","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.organomet.4c00359","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
引用次数: 0
Abstract
Cationic gold-alkyne chemistry is a vital component of homogeneous gold catalysis due to its ability to access a wide range of reactive intermediates. Direct C(sp3)–H insertion by the cationic gold-alkyne complex is an emergent reaction in this area without a well-defined reactive intermediate. Gold(I)-catalyzed isomerization of cyclooctyne facilitated by trans-annular C(sp3)–H insertion ( Eur. J. Inorg. Chem.2016, 995−1001) is used as a case study to investigate the mechanism of this process with density functional theory (DFT) calculations. Natural resonance theory (NRT) calculations are used to analyze the reactive intermediate in terms of familiar resonance structures. It is found that “slippage” or deformation of the gold ion coordination from η2 to η1 increases the NRT weighting of the vinyl cation resonance structure from 12% to 25% leading to its C(sp3)–H insertion reactivity. In addition, DFT calculations and a distortion-interaction analysis are used to rationalize the catalyst-dependent regioselectivity observed in the reaction. Lastly, model studies investigating the impact of the alkyne substrate and ancillary ligand show that electron-withdrawing substituents and electron deficient ligands lower Gibbs activation energy for C(sp3)–H insertion, which suggests strategies to further improve the reaction through catalyst design.
期刊介绍:
Organometallics is the flagship journal of organometallic chemistry and records progress in one of the most active fields of science, bridging organic and inorganic chemistry. The journal publishes Articles, Communications, Reviews, and Tutorials (instructional overviews) that depict research on the synthesis, structure, bonding, chemical reactivity, and reaction mechanisms for a variety of applications, including catalyst design and catalytic processes; main-group, transition-metal, and lanthanide and actinide metal chemistry; synthetic aspects of polymer science and materials science; and bioorganometallic chemistry.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
