Junrong Song , Zhiming Song , Yuanli Gong , Lichang Ge , Wenlu Lou
{"title":"Advancing cancer driver gene identification through an integrative network and pathway approach","authors":"Junrong Song , Zhiming Song , Yuanli Gong , Lichang Ge , Wenlu Lou","doi":"10.1016/j.jbi.2024.104729","DOIUrl":null,"url":null,"abstract":"<div><h3>Objective</h3><div>Cancer is a complex genetic disease characterized by the accumulation of various mutations, with driver genes playing a crucial role in cancer initiation and progression. Distinguishing driver genes from passenger mutations is essential for understanding cancer biology and discovering therapeutic targets. However, the majority of existing methods ignore the mutational heterogeneity and commonalities among patients, which hinders the identification of driver genes more effectively.</div></div><div><h3>Methods</h3><div>This study introduces MCSdriver, a novel computational model that integrates network and pathway information to prioritize the identification of cancer driver genes. MCSdriver employs a bidirectional random walk algorithm to quantify the mutual exclusivity and functional relationships between mutated genes within patient cohorts. It calculates similarity scores based on a mutual exclusivity-weighted network and pathway coverage patterns, accounting for patient-specific heterogeneity and molecular profile similarity.</div></div><div><h3>Results</h3><div>This approach enhances the accuracy and quality of driver gene identification. MCSdriver demonstrates superior performance in identifying cancer driver genes across four cancer types from The Cancer Genome Atlas, showing a higher F-score, Recall and Precision compared to existing ranking list-based and module-based models.</div></div><div><h3>Conclusion</h3><div>The MCSdriver model not only outperforms other models in identifying known cancer driver genes but also effectively identifies novel driver genes involved in cancer-related biological processes. The model’s consideration of patient-specific heterogeneity and similarity in molecular profiles significantly enhances the accuracy and quality of driver gene identification. Validation through Gene Ontology enrichment analysis and literature mining further underscores its potential application value in personalized cancer therapy, offering a promising tool for advancing our understanding and treatment of cancer.</div></div>","PeriodicalId":15263,"journal":{"name":"Journal of Biomedical Informatics","volume":"158 ","pages":"Article 104729"},"PeriodicalIF":4.5000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Biomedical Informatics","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1532046424001473","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/19 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS","Score":null,"Total":0}
引用次数: 0
Abstract
Objective
Cancer is a complex genetic disease characterized by the accumulation of various mutations, with driver genes playing a crucial role in cancer initiation and progression. Distinguishing driver genes from passenger mutations is essential for understanding cancer biology and discovering therapeutic targets. However, the majority of existing methods ignore the mutational heterogeneity and commonalities among patients, which hinders the identification of driver genes more effectively.
Methods
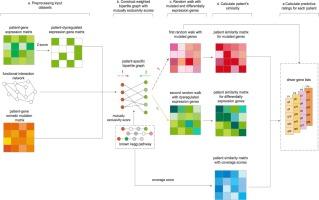
This study introduces MCSdriver, a novel computational model that integrates network and pathway information to prioritize the identification of cancer driver genes. MCSdriver employs a bidirectional random walk algorithm to quantify the mutual exclusivity and functional relationships between mutated genes within patient cohorts. It calculates similarity scores based on a mutual exclusivity-weighted network and pathway coverage patterns, accounting for patient-specific heterogeneity and molecular profile similarity.
Results
This approach enhances the accuracy and quality of driver gene identification. MCSdriver demonstrates superior performance in identifying cancer driver genes across four cancer types from The Cancer Genome Atlas, showing a higher F-score, Recall and Precision compared to existing ranking list-based and module-based models.
Conclusion
The MCSdriver model not only outperforms other models in identifying known cancer driver genes but also effectively identifies novel driver genes involved in cancer-related biological processes. The model’s consideration of patient-specific heterogeneity and similarity in molecular profiles significantly enhances the accuracy and quality of driver gene identification. Validation through Gene Ontology enrichment analysis and literature mining further underscores its potential application value in personalized cancer therapy, offering a promising tool for advancing our understanding and treatment of cancer.
期刊介绍:
The Journal of Biomedical Informatics reflects a commitment to high-quality original research papers, reviews, and commentaries in the area of biomedical informatics methodology. Although we publish articles motivated by applications in the biomedical sciences (for example, clinical medicine, health care, population health, and translational bioinformatics), the journal emphasizes reports of new methodologies and techniques that have general applicability and that form the basis for the evolving science of biomedical informatics. Articles on medical devices; evaluations of implemented systems (including clinical trials of information technologies); or papers that provide insight into a biological process, a specific disease, or treatment options would generally be more suitable for publication in other venues. Papers on applications of signal processing and image analysis are often more suitable for biomedical engineering journals or other informatics journals, although we do publish papers that emphasize the information management and knowledge representation/modeling issues that arise in the storage and use of biological signals and images. System descriptions are welcome if they illustrate and substantiate the underlying methodology that is the principal focus of the report and an effort is made to address the generalizability and/or range of application of that methodology. Note also that, given the international nature of JBI, papers that deal with specific languages other than English, or with country-specific health systems or approaches, are acceptable for JBI only if they offer generalizable lessons that are relevant to the broad JBI readership, regardless of their country, language, culture, or health system.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
