Josianne Nunes Carriço, Catarina Inês Gonçalves, Asma Al-Naama, Najeeb Syed, José Maria Aragüés, Margarida Bastos, Fernando Fonseca, Teresa Borges, Bernardo Dias Pereira, Duarte Pignatelli, Davide Carvalho, Filipe Cunha, Ana Saavedra, Elisabete Rodrigues, Joana Saraiva, Luisa Ruas, Nuno Vicente, João Martin Martins, Adriana De Sousa Lages, Maria João Oliveira, Cíntia Castro-Correia, Miguel Melo, Raquel Gomes Martins, Joana Couto, Carolina Moreno, Diana Martins, Patrícia Oliveira, Teresa Martins, Sofia Almeida Martins, Olinda Marques, Carla Meireles, António Garrão, Cláudia Nogueira, Carla Baptista, Susana Gama-de-Sousa, Cláudia Amaral, Mariana Martinho, Catarina Limbert, Luisa Barros, Inês Henriques Vieira, Teresa Sabino, Luís R Saraiva, Manuel Carlos Lemos
{"title":"Genetic architecture of congenital hypogonadotropic hypogonadism: insights from analysis of a Portuguese cohort.","authors":"Josianne Nunes Carriço, Catarina Inês Gonçalves, Asma Al-Naama, Najeeb Syed, José Maria Aragüés, Margarida Bastos, Fernando Fonseca, Teresa Borges, Bernardo Dias Pereira, Duarte Pignatelli, Davide Carvalho, Filipe Cunha, Ana Saavedra, Elisabete Rodrigues, Joana Saraiva, Luisa Ruas, Nuno Vicente, João Martin Martins, Adriana De Sousa Lages, Maria João Oliveira, Cíntia Castro-Correia, Miguel Melo, Raquel Gomes Martins, Joana Couto, Carolina Moreno, Diana Martins, Patrícia Oliveira, Teresa Martins, Sofia Almeida Martins, Olinda Marques, Carla Meireles, António Garrão, Cláudia Nogueira, Carla Baptista, Susana Gama-de-Sousa, Cláudia Amaral, Mariana Martinho, Catarina Limbert, Luisa Barros, Inês Henriques Vieira, Teresa Sabino, Luís R Saraiva, Manuel Carlos Lemos","doi":"10.1093/hropen/hoae053","DOIUrl":null,"url":null,"abstract":"<p><strong>Study question: </strong>What is the contribution of genetic defects in Portuguese patients with congenital hypogonadotropic hypogonadism (CHH)?</p><p><strong>Summary answer: </strong>Approximately one-third of patients with CHH were found to have a genetic cause for their disorder, with causal pathogenic and likely pathogenic germline variants distributed among 10 different genes; cases of oligogenic inheritance were also included.</p><p><strong>What is known already: </strong>CHH is a rare and genetically heterogeneous disorder characterized by deficient production, secretion, or action of GnRH, LH, and FSH, resulting in delayed or absent puberty, and infertility.</p><p><strong>Study design size duration: </strong>Genetic screening was performed on a cohort of 81 Portuguese patients with CHH (36 with Kallmann syndrome and 45 with normosmic hypogonadotropic hypogonadism) and 263 unaffected controls.</p><p><strong>Participants/materials setting methods: </strong>The genetic analysis was performed by whole-exome sequencing followed by the analysis of a virtual panel of 169 CHH-associated genes. The main outcome measures were non-synonymous rare sequence variants (population allele frequency <0.01) classified as pathogenic, likely pathogenic, and variants of uncertain significance (VUS).</p><p><strong>Main results and the role of chance: </strong>A genetic cause was identified in 29.6% of patients. Causal pathogenic and likely pathogenic variants were distributed among 10 of the analysed genes. The most frequently implicated genes were <i>GNRHR</i>, <i>FGFR1</i>, <i>ANOS1</i>, and <i>CHD7</i>. Oligogenicity for pathogenic and likely pathogenic variants was observed in 6.2% of patients. VUS and oligogenicity for VUS variants were observed in 85.2% and 54.3% of patients, respectively, but were not significantly different from that observed in controls.</p><p><strong>Large scale data: </strong>N/A.</p><p><strong>Limitations reasons for caution: </strong>The identification of a large number of VUS presents challenges in interpretation and these may require reclassification as more evidence becomes available. Non-coding and copy number variants were not studied. Functional studies of the variants were not undertaken.</p><p><strong>Wider implications of the findings: </strong>This study highlights the genetic heterogeneity of CHH and identified several novel variants that expand the mutational spectrum of the disorder. A significant proportion of patients remained without a genetic diagnosis, suggesting the involvement of additional genetic, epigenetic, or environmental factors. The high frequency of VUS underscores the importance of cautious variant interpretation. These findings contribute to the understanding of the genetic architecture of CHH and emphasize the need for further studies to elucidate the underlying mechanisms and identify additional causes of CHH.</p><p><strong>Study funding/competing interests: </strong>This research was funded by the Portuguese Foundation for Science and Technology (grant numbers PTDC/SAU-GMG/098419/2008, UIDB/00709/2020, CEECINST/00016/2021/CP2828/CT0002, and 2020.04924.BD) and by Sidra Medicine-a member of the Qatar Foundation (grant number SDR400038). The authors declare no competing interests.</p>","PeriodicalId":73264,"journal":{"name":"Human reproduction open","volume":"2024 3","pages":"hoae053"},"PeriodicalIF":11.1000,"publicationDate":"2024-09-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11415827/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human reproduction open","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/hropen/hoae053","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"OBSTETRICS & GYNECOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Study question: What is the contribution of genetic defects in Portuguese patients with congenital hypogonadotropic hypogonadism (CHH)?
Summary answer: Approximately one-third of patients with CHH were found to have a genetic cause for their disorder, with causal pathogenic and likely pathogenic germline variants distributed among 10 different genes; cases of oligogenic inheritance were also included.
What is known already: CHH is a rare and genetically heterogeneous disorder characterized by deficient production, secretion, or action of GnRH, LH, and FSH, resulting in delayed or absent puberty, and infertility.
Study design size duration: Genetic screening was performed on a cohort of 81 Portuguese patients with CHH (36 with Kallmann syndrome and 45 with normosmic hypogonadotropic hypogonadism) and 263 unaffected controls.
Participants/materials setting methods: The genetic analysis was performed by whole-exome sequencing followed by the analysis of a virtual panel of 169 CHH-associated genes. The main outcome measures were non-synonymous rare sequence variants (population allele frequency <0.01) classified as pathogenic, likely pathogenic, and variants of uncertain significance (VUS).
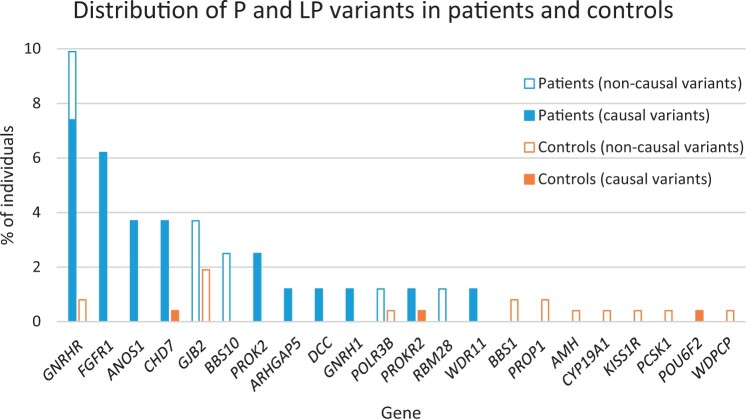
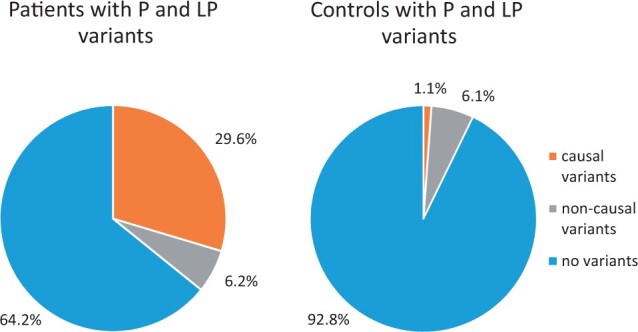
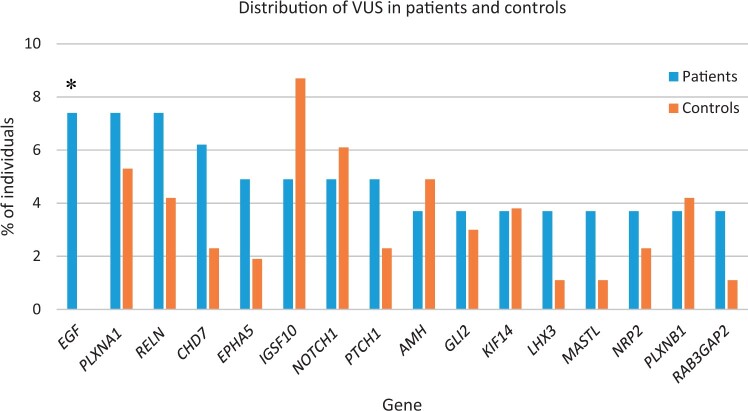
Main results and the role of chance: A genetic cause was identified in 29.6% of patients. Causal pathogenic and likely pathogenic variants were distributed among 10 of the analysed genes. The most frequently implicated genes were GNRHR, FGFR1, ANOS1, and CHD7. Oligogenicity for pathogenic and likely pathogenic variants was observed in 6.2% of patients. VUS and oligogenicity for VUS variants were observed in 85.2% and 54.3% of patients, respectively, but were not significantly different from that observed in controls.
Large scale data: N/A.
Limitations reasons for caution: The identification of a large number of VUS presents challenges in interpretation and these may require reclassification as more evidence becomes available. Non-coding and copy number variants were not studied. Functional studies of the variants were not undertaken.
Wider implications of the findings: This study highlights the genetic heterogeneity of CHH and identified several novel variants that expand the mutational spectrum of the disorder. A significant proportion of patients remained without a genetic diagnosis, suggesting the involvement of additional genetic, epigenetic, or environmental factors. The high frequency of VUS underscores the importance of cautious variant interpretation. These findings contribute to the understanding of the genetic architecture of CHH and emphasize the need for further studies to elucidate the underlying mechanisms and identify additional causes of CHH.
Study funding/competing interests: This research was funded by the Portuguese Foundation for Science and Technology (grant numbers PTDC/SAU-GMG/098419/2008, UIDB/00709/2020, CEECINST/00016/2021/CP2828/CT0002, and 2020.04924.BD) and by Sidra Medicine-a member of the Qatar Foundation (grant number SDR400038). The authors declare no competing interests.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
