{"title":"Excess glucose alone induces hepatocyte damage due to oxidative stress and endoplasmic reticulum stress","authors":"Tsuguru Hayashi, Shinji Oe, Koichiro Miyagawa, Masashi Kusanaga, Noriyoshi Ogino, Yuichi Honma, Masaru Harada","doi":"10.1016/j.yexcr.2024.114264","DOIUrl":null,"url":null,"abstract":"<div><div>Type 2 diabetes mellitus (DM) is a significant risk factor for metabolic dysfunction-associated steatotic liver disease (MASLD) and hepatocellular carcinoma (HCC). With the increasing prevalence of type 2 DM and MASLD due to lifestyle changes, understanding their impact on liver health is crucial. However, the hepatocellular damage caused by glucose alone is unknown. This study investigates the effect of excess glucose on hepatocytes, focusing on oxidative stress, endoplasmic reticulum stress (ER stress), apoptosis, autophagy, and cell proliferation. We treated an immortalized-human hepatocyte cell line with excess glucose and analyzed. Excess glucose induced oxidative stress and ER stress in a time- and concentration-dependent manner, leading to apoptosis. Oxidative stress and ER stress were independently induced by excess glucose. Proteasome inhibitors and palmitic acid exacerbated glucose-induced stress, leading to the formation of Mallory-Denk body-like inclusion bodies. Despite these stresses, autophagic flux was not altered. Excess glucose also caused DNA damage but did not affect cell proliferation. This suggests that glucose itself can contribute to the progression of metabolic dysfunction-associated steatohepatitis (MASH) and carcinogenesis of HCC in patients with type 2 DM. Managing blood glucose levels is crucial to prevent hepatocyte damage and associated complications.</div></div>","PeriodicalId":12227,"journal":{"name":"Experimental cell research","volume":"442 2","pages":"Article 114264"},"PeriodicalIF":3.5000,"publicationDate":"2024-09-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Experimental cell research","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0014482724003550","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
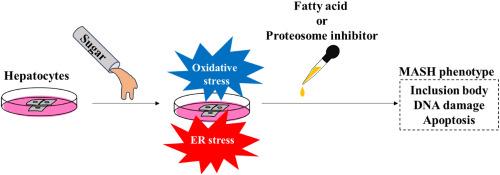
Type 2 diabetes mellitus (DM) is a significant risk factor for metabolic dysfunction-associated steatotic liver disease (MASLD) and hepatocellular carcinoma (HCC). With the increasing prevalence of type 2 DM and MASLD due to lifestyle changes, understanding their impact on liver health is crucial. However, the hepatocellular damage caused by glucose alone is unknown. This study investigates the effect of excess glucose on hepatocytes, focusing on oxidative stress, endoplasmic reticulum stress (ER stress), apoptosis, autophagy, and cell proliferation. We treated an immortalized-human hepatocyte cell line with excess glucose and analyzed. Excess glucose induced oxidative stress and ER stress in a time- and concentration-dependent manner, leading to apoptosis. Oxidative stress and ER stress were independently induced by excess glucose. Proteasome inhibitors and palmitic acid exacerbated glucose-induced stress, leading to the formation of Mallory-Denk body-like inclusion bodies. Despite these stresses, autophagic flux was not altered. Excess glucose also caused DNA damage but did not affect cell proliferation. This suggests that glucose itself can contribute to the progression of metabolic dysfunction-associated steatohepatitis (MASH) and carcinogenesis of HCC in patients with type 2 DM. Managing blood glucose levels is crucial to prevent hepatocyte damage and associated complications.
期刊介绍:
Our scope includes but is not limited to areas such as: Chromosome biology; Chromatin and epigenetics; DNA repair; Gene regulation; Nuclear import-export; RNA processing; Non-coding RNAs; Organelle biology; The cytoskeleton; Intracellular trafficking; Cell-cell and cell-matrix interactions; Cell motility and migration; Cell proliferation; Cellular differentiation; Signal transduction; Programmed cell death.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
