Evaluation of inhibition effect and interaction mechanism of antiviral drugs on main protease of novel coronavirus: Molecular docking and molecular dynamics studies
Xin Gao , Cuihong Wang , Yue Jiang , Shouchao Zhang , Meiling Zhang , Lijuan Liu , Sendan Gao
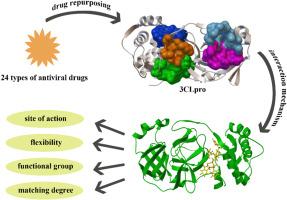
{"title":"Evaluation of inhibition effect and interaction mechanism of antiviral drugs on main protease of novel coronavirus: Molecular docking and molecular dynamics studies","authors":"Xin Gao , Cuihong Wang , Yue Jiang , Shouchao Zhang , Meiling Zhang , Lijuan Liu , Sendan Gao","doi":"10.1016/j.jmgm.2024.108873","DOIUrl":null,"url":null,"abstract":"<div><div>The outbreak of pneumonia caused by the novel coronavirus (SARS-CoV-2) has presented a challenge to public health. The identification and development of effective antiviral drugs is essential. The main protease (3CLpro) plays an important role in the viral replication of SARS-CoV-2 and is considered to be an effective therapeutic target. In this study, according to the principle of drug repurposing, a variety of antiviral drugs commonly used were studied by molecular docking and molecular dynamics (MD) simulations to obtain potential inhibitors of main proteases. 24 antiviral drugs were docked with 5 potential action sites of 3CLpro, and the drugs with high binding strength were further simulated by MD and the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) binding free energy calculations. The results showed that the drugs with high flexibility could bind to 3CLpro better than those with low flexibility. The interaction mechanism between antiviral drugs and main protease was analyzed in detail by calculating the root mean square displacement (RMSD), root mean square fluctuation (RMSF) and interaction residues properties. The results showed that the six drugs with high flexibility (Remdesivir, Simnotrelvir, Sofosbuvir, Ledipasvir, Indinavir and Raltegravir) had strong binding strength with 3CLpro, and the last four antiviral drugs can be used as potential candidates for main protease inhibitors.</div></div>","PeriodicalId":16361,"journal":{"name":"Journal of molecular graphics & modelling","volume":"133 ","pages":"Article 108873"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular graphics & modelling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1093326324001736","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/24 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
The outbreak of pneumonia caused by the novel coronavirus (SARS-CoV-2) has presented a challenge to public health. The identification and development of effective antiviral drugs is essential. The main protease (3CLpro) plays an important role in the viral replication of SARS-CoV-2 and is considered to be an effective therapeutic target. In this study, according to the principle of drug repurposing, a variety of antiviral drugs commonly used were studied by molecular docking and molecular dynamics (MD) simulations to obtain potential inhibitors of main proteases. 24 antiviral drugs were docked with 5 potential action sites of 3CLpro, and the drugs with high binding strength were further simulated by MD and the molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) binding free energy calculations. The results showed that the drugs with high flexibility could bind to 3CLpro better than those with low flexibility. The interaction mechanism between antiviral drugs and main protease was analyzed in detail by calculating the root mean square displacement (RMSD), root mean square fluctuation (RMSF) and interaction residues properties. The results showed that the six drugs with high flexibility (Remdesivir, Simnotrelvir, Sofosbuvir, Ledipasvir, Indinavir and Raltegravir) had strong binding strength with 3CLpro, and the last four antiviral drugs can be used as potential candidates for main protease inhibitors.
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
