N. Ngom , A.J. Etindele , N.F. Andriambelaza , C. Nithaya , A.S. Wakata , S. Kenmoe
{"title":"Comparative study of CO adsorption on Au, Cu, MoO2 and MoS2 2D Nanoparticles","authors":"N. Ngom , A.J. Etindele , N.F. Andriambelaza , C. Nithaya , A.S. Wakata , S. Kenmoe","doi":"10.1016/j.comptc.2024.114877","DOIUrl":null,"url":null,"abstract":"<div><div>This study focuses on a comparative analysis of the electronic properties of triangular, irregular hexagonal, and octagonal 2D nanoparticles containing 10, 12, and 14 motifs, made of Au, Cu, MoO<sub>2</sub>, and MoS<sub>2</sub>. The investigation was carried out using density functional theory. The formation energies and vibrational frequencies demonstrate that the 2D nanostructure configurations can exist as stable structures. Edge atoms with lower coordination numbers than central atoms, are the preferred sites for CO adsorption. Using orbital-weighting dual descriptors calculated from Fukui functions enabled the identification of a majority nucleophilic attack sites in Au and Cu nanoparticles, while MoO<sub>2</sub> and MoS<sub>2</sub>-based nanoparticles present almost as many electrophilic sites as nucleophilic sites. The charge transferred between the nanostructures and the CO molecule and the redistribution of the projected density of states were used to assess the strength of interfacial bonds and the nature of the fundamental interaction involved in the bonding.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1241 ","pages":"Article 114877"},"PeriodicalIF":3.0000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X2400416X","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/18 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
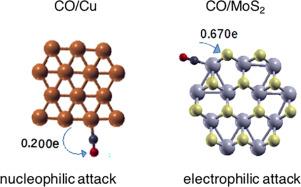
This study focuses on a comparative analysis of the electronic properties of triangular, irregular hexagonal, and octagonal 2D nanoparticles containing 10, 12, and 14 motifs, made of Au, Cu, MoO2, and MoS2. The investigation was carried out using density functional theory. The formation energies and vibrational frequencies demonstrate that the 2D nanostructure configurations can exist as stable structures. Edge atoms with lower coordination numbers than central atoms, are the preferred sites for CO adsorption. Using orbital-weighting dual descriptors calculated from Fukui functions enabled the identification of a majority nucleophilic attack sites in Au and Cu nanoparticles, while MoO2 and MoS2-based nanoparticles present almost as many electrophilic sites as nucleophilic sites. The charge transferred between the nanostructures and the CO molecule and the redistribution of the projected density of states were used to assess the strength of interfacial bonds and the nature of the fundamental interaction involved in the bonding.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
