{"title":"Exploring the Computational Aspects of Propylene Oligomerization Catalysis Using M′2M Type Trimetallic MOF Nodes","authors":"Rishu Khurana, Valay Agarawal, Cong Liu","doi":"10.1021/acs.jpcc.4c04992","DOIUrl":null,"url":null,"abstract":"Metal–organic frameworks have emerged as promising materials in the field of catalysis. They offer an optimal ground for screening catalysts and tailoring their catalytic properties. In this work, via density functional theory (DFT) calculations, we investigated the catalytic activity of the trimetallic MOF nodes, M′<sub>2</sub>M for propylene oligomerization, by varying the active metal M from Sc to Cu with M′ being Fe, aiming to grasp the impact of altering the active atoms on the catalyst’s activity. Additionally, we examined how substituting the spectator atom, M′, with other transition metals, i.e., from Sc to Cu, affects these energy barriers, keeping Ni as the active metal. We proposed several cases with lower or comparable energy barriers to the experimentally reported Fe<sub>2</sub>Ni trimetallic MOF node. In addition, we found a correlative relationship between spin-density from natural population analysis and energy barriers in the realm of C–C bond formation, whereby an elevation in spin-density is found to be inversely proportional to the magnitude of the energy barriers. Moreover, we calculated the energy barriers for C–C coupling and β-hydride elimination using multireference NEVPT2 calculations on top of the CASSCF wave function to validate the rate-determining step that is predicted by DFT.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"39 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2024-09-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.4c04992","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
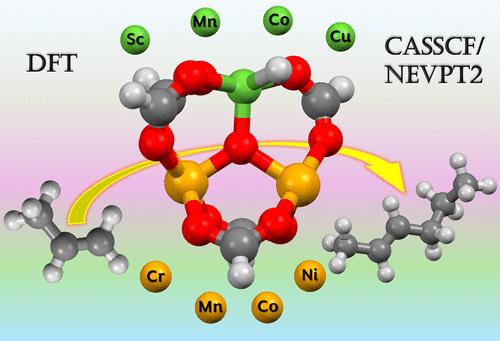
Abstract
Metal–organic frameworks have emerged as promising materials in the field of catalysis. They offer an optimal ground for screening catalysts and tailoring their catalytic properties. In this work, via density functional theory (DFT) calculations, we investigated the catalytic activity of the trimetallic MOF nodes, M′2M for propylene oligomerization, by varying the active metal M from Sc to Cu with M′ being Fe, aiming to grasp the impact of altering the active atoms on the catalyst’s activity. Additionally, we examined how substituting the spectator atom, M′, with other transition metals, i.e., from Sc to Cu, affects these energy barriers, keeping Ni as the active metal. We proposed several cases with lower or comparable energy barriers to the experimentally reported Fe2Ni trimetallic MOF node. In addition, we found a correlative relationship between spin-density from natural population analysis and energy barriers in the realm of C–C bond formation, whereby an elevation in spin-density is found to be inversely proportional to the magnitude of the energy barriers. Moreover, we calculated the energy barriers for C–C coupling and β-hydride elimination using multireference NEVPT2 calculations on top of the CASSCF wave function to validate the rate-determining step that is predicted by DFT.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
