XiaoYing Sun, Jianzhuo Lu, Bin Qin, Zhen Zhao, Bo Li
{"title":"Revealing of Intrinsic Relation between Surface Atomic Geometry and Catalytic Mechanism of CuNi Alloy Catalyst in CO2 Hydrogenation to Methanol","authors":"XiaoYing Sun, Jianzhuo Lu, Bin Qin, Zhen Zhao, Bo Li","doi":"10.1021/acs.jpcc.4c01438","DOIUrl":null,"url":null,"abstract":"It is well-known that the catalytic mechanism and performance are strongly dependent on the exposed surfaces of metal catalysts. This concept is further broadened and elaborated in the current work for the bimetallic NiCu catalyst in CO<sub>2</sub> hydrogenation to methanol. (100), (110), and (111) facets of NiCu with either Cu or Ni termination, which consisted of five different surfaces, are investigated by DFT-based microkinetic simulation. The unique surface geometry of the alloy surface results in the distinct electronic structure as revealed from electron localization function and PDOS analyses. CO<sub>2</sub> molecule exhibits strong chemisorption on all surfaces with a bent bond angle which is significantly activated upon adsorption. Three mechanisms, including formate, RWGS + COhydro, and CO + O, are explored at the same footing. It is concluded that the first step in the pathway has pivotal importance to determine the favorable mechanism. From the reaction pathway analysis, it is found that direct C–O bond breaking has a comparable barrier with the hydrogenation step which is not usually observed for metallic catalysts in CO<sub>2</sub> hydrogenation. In general, hydrogen addition to carbon in the CO<sub>2</sub> molecule has a smaller barrier than the counterpart of addition to oxygen. The precisely exposed atom on the bimetallic catalyst largely determined both performance and mechanism which complemented the concept of surface-dependent catalytic properties. Microkinetic simulation indicated that the (111) surface with Cu termination has the largest TOF of methanol formation in the experimental relevant temperature, and the favorable mechanism is ascribed to be formate. Moreover, both thermodynamics and kinetic control steps are identified from the degree of rate control analysis. Overall, the current work deepened the importance of fine tuning the surface geometry to adjust the catalytic performance and mechanism and paved the way for further development of bimetallic catalysts for CO<sub>2</sub> hydrogenation.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"45 1","pages":""},"PeriodicalIF":3.2000,"publicationDate":"2024-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.4c01438","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
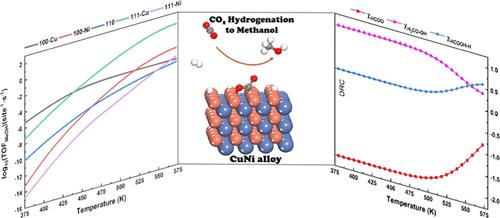
It is well-known that the catalytic mechanism and performance are strongly dependent on the exposed surfaces of metal catalysts. This concept is further broadened and elaborated in the current work for the bimetallic NiCu catalyst in CO2 hydrogenation to methanol. (100), (110), and (111) facets of NiCu with either Cu or Ni termination, which consisted of five different surfaces, are investigated by DFT-based microkinetic simulation. The unique surface geometry of the alloy surface results in the distinct electronic structure as revealed from electron localization function and PDOS analyses. CO2 molecule exhibits strong chemisorption on all surfaces with a bent bond angle which is significantly activated upon adsorption. Three mechanisms, including formate, RWGS + COhydro, and CO + O, are explored at the same footing. It is concluded that the first step in the pathway has pivotal importance to determine the favorable mechanism. From the reaction pathway analysis, it is found that direct C–O bond breaking has a comparable barrier with the hydrogenation step which is not usually observed for metallic catalysts in CO2 hydrogenation. In general, hydrogen addition to carbon in the CO2 molecule has a smaller barrier than the counterpart of addition to oxygen. The precisely exposed atom on the bimetallic catalyst largely determined both performance and mechanism which complemented the concept of surface-dependent catalytic properties. Microkinetic simulation indicated that the (111) surface with Cu termination has the largest TOF of methanol formation in the experimental relevant temperature, and the favorable mechanism is ascribed to be formate. Moreover, both thermodynamics and kinetic control steps are identified from the degree of rate control analysis. Overall, the current work deepened the importance of fine tuning the surface geometry to adjust the catalytic performance and mechanism and paved the way for further development of bimetallic catalysts for CO2 hydrogenation.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
