Multistep screening of transition-metal-based homonuclear double-atom catalysts to unravel the electronic origin of their activity and selectivity challenges for nitrogen reduction†
{"title":"Multistep screening of transition-metal-based homonuclear double-atom catalysts to unravel the electronic origin of their activity and selectivity challenges for nitrogen reduction†","authors":"Anjumun Rasool , Manzoor Ahmad Dar","doi":"10.1039/d4cy00480a","DOIUrl":null,"url":null,"abstract":"<div><div>Lack of robust catalyst design strategies for tackling the selectivity and activity challenges poses serious limitations in the development of efficient catalysts for nitrogen reduction to ammonia. The synergistic interactions in double-atom catalysts (DACs) have aroused great interest in developing promising catalytic centers for the nitrogen reduction reaction (NRR). Using a multistep screening strategy based on systematic first-principles simulations, we find that Fe<sub>2</sub>, Co<sub>2</sub>, and W<sub>2</sub> dimer species impregnated in a tetracyanoquinodimethane-based monolayer achieve suitable adsorption behaviour for the various NRR intermediates, leading to excellent activity and selectivity among the 27 DACs considered in this study for the NRR. Interestingly, our results reveal very low limiting potential values of −0.56, −0.58, and −0.53 V for Fe<sub>2</sub>, Co<sub>2</sub>, and W<sub>2</sub>, respectively, compared to the experimentally reported values of −0.73 and −0.98 V for the Ru-based single-atom catalyst and Ru(0001) stepped surface. Density of states analysis indicated that the adsorption pattern of the reaction intermediates was regulated by the d-states of the DACs near the Fermi level. Correlation trends between the limiting potential and the free energy change for adsorption of different intermediates show that the free energy change for N<sub>2</sub> adsorption proves a suitable guidance to evaluate the NRR activity of the modelled catalysts. Further, rigorous electronic structure analysis highlighted properties such as integrated crystal orbital Hamilton populations and orbital projected density of states, and the d-band centre could be successfully used to rationalize the N<sub>2</sub> binding and adsorption on these catalysts. Thus, this work provides a feasible design strategy for NRR electrocatalysis based on extensive electronic structure concepts.</div></div>","PeriodicalId":66,"journal":{"name":"Catalysis Science & Technology","volume":"14 19","pages":"Pages 5687-5698"},"PeriodicalIF":4.2000,"publicationDate":"2024-09-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Catalysis Science & Technology","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/org/science/article/pii/S2044475324004854","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/2 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
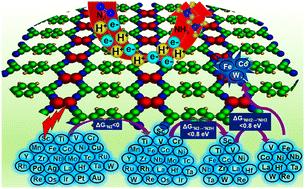
Lack of robust catalyst design strategies for tackling the selectivity and activity challenges poses serious limitations in the development of efficient catalysts for nitrogen reduction to ammonia. The synergistic interactions in double-atom catalysts (DACs) have aroused great interest in developing promising catalytic centers for the nitrogen reduction reaction (NRR). Using a multistep screening strategy based on systematic first-principles simulations, we find that Fe2, Co2, and W2 dimer species impregnated in a tetracyanoquinodimethane-based monolayer achieve suitable adsorption behaviour for the various NRR intermediates, leading to excellent activity and selectivity among the 27 DACs considered in this study for the NRR. Interestingly, our results reveal very low limiting potential values of −0.56, −0.58, and −0.53 V for Fe2, Co2, and W2, respectively, compared to the experimentally reported values of −0.73 and −0.98 V for the Ru-based single-atom catalyst and Ru(0001) stepped surface. Density of states analysis indicated that the adsorption pattern of the reaction intermediates was regulated by the d-states of the DACs near the Fermi level. Correlation trends between the limiting potential and the free energy change for adsorption of different intermediates show that the free energy change for N2 adsorption proves a suitable guidance to evaluate the NRR activity of the modelled catalysts. Further, rigorous electronic structure analysis highlighted properties such as integrated crystal orbital Hamilton populations and orbital projected density of states, and the d-band centre could be successfully used to rationalize the N2 binding and adsorption on these catalysts. Thus, this work provides a feasible design strategy for NRR electrocatalysis based on extensive electronic structure concepts.
期刊介绍:
A multidisciplinary journal focusing on cutting edge research across all fundamental science and technological aspects of catalysis.
Editor-in-chief: Bert Weckhuysen
Impact factor: 5.0
Time to first decision (peer reviewed only): 31 days



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
